
王风平 , 陈云如
, 陈云如
Wang Fengping, Chen Yunru
中图分类号: P735
文献标识码: A
文章编号: 1001-8166(2017)12-1277-10
收稿日期: 2017-10-16
修回日期: 2017-11-28
网络出版日期: 2017-12-20
版权声明: 2017 地球科学进展 编辑部
基金资助:
作者简介:
First author:Wang Fengping(1971-),female,Danjiangkou City,Hubei Province,Professor. Research areas include deep biosphere.E-mail:fengpingw@sjtu.edu.cn
作者简介:王风平(1971-),女,湖北丹江口人,教授,主要从事深部生物圈研究.E-mail:fengpingw@stju.edu.cn
展开
摘要
发现海底沉积物深处乃至岩石中仍然有生命,是“大洋钻探计划”开展近半个世纪以来最令人激动的重要发现之一,即大洋“深部生物圈”的发现。近年来,深部生物圈的发现与探索已成为地质学和生物学领域最令人兴奋的研究前沿之一,生物圈前沿即深部生命、生物多样性和环境驱动的生态系统成为新的国际大洋发现计划(IODP 2013-2023)的四大研究主题之一。通过对深部生物圈研究历史进行简单回顾,介绍深部生物圈及其环境特征,目前已经完成的以探究地壳和海洋沉积物中生物圈为目标的大洋钻探计划(IODP)航次,深部生物圈研究的前沿科学问题,已经取得的重大研究进展和面临的挑战,以及中国科学家的贡献和深部生物圈的未来发展的建议与展望。
关键词:
Abstract
The discovery of living microorganisms deep in the marine sediments and even in the oceanic crust (the marine “deep biosphere”), is one of the most significant and exciting discoveries since the ocean drilling program began almost a half-century ago. Investigation of the deep biosphere has become the most thrilling research frontier for both geological and biological sciences. The “biosphere frontiers” has been listed as one of the four themes in the 10-year plan of the International Ocean Discovery Program (IODP 2012-2023), including deep life, biodiversity and environmental forcing of ecosystems. Here, we introduced the deep biosphere and its environmental features, several completed Integrated Ocean Drilling Program Expeditions, which targeted the subseafloor deep biosphere within the crust and sediments, and highlighted the main progress we have made in deep biosphere and deep life research, especially the contribution of Chinese scientists. Finally, we will give a perspective on the future of deep biosphere research according to the challenge we are facing and the key questions need to be answered.
Keywords:
深部生物圈即埋藏在地球深部的黑暗生态系统,一般指海底沉积物1 m以下环境中栖息的生态系统[1]。不同于利用太阳能进行光合作用维持的地表生物圈,深部微生物可通过化能合成作用将无机物转化为有机质,并作为初级生产者为消费者提供能量和碳源,形成一个“黑暗食物链”。对于黑暗海底生命的探索可以追溯到20世纪30年代,美国Scripps海洋研究所的ZoBell等[2]发现在海床下数厘米至数米范围内的深海沉积物内存在细菌,开始观测支持这个深部生物圈(subsurface biosphere)的产能过程[3,4]。1977年, 美国“Alvin”号深潜器首次在太平洋的加拉帕戈斯(Galapagos)洋中脊发现了深海热液喷口及其周围的热液生态系统[5]。Parkes等[6]通过大洋钻探, 在海床下数百米深的沉积物样品中发现了具有活性的微生物。这些早期偶然性的海底勘测驱使人们对于海底生物圈开展了更有计划的系统性探测, 从而发现了这个“隐藏”在地球内部的巨大的生命栖息之所[7]。
海洋约覆盖了地球表面积的70%,海水以下存在大量形态各异的生态环境,例如,海洋沉积物、洋壳、热液喷口和冷泉等,这些环境构成了地球上的生物,尤其是微生物最大的栖息地[8]。深部生物圈包括海洋和陆地,本文仅介绍海洋深部生物圈,主要指在海床以下沉积物和岩石以及其中的流体中生活的生物统称,其中热液喷口和冷泉被认为是地球深部和上层海洋连接的通道,是研究深部生物圈的理想窗口。深部生物圈虽然深埋海底,却不是一个孤立的系统,而是与水圈有着非常紧密的联系,据估算每20万年相当于全球海水体积的流体会通过洋壳含水层循环一次[9],极有可能在全球尺度上影响生物地球化学过程,包括碳循环、营养物质循环、能量流以及气候。因此,对深部生物圈的研究不仅吸引了科学家探索未知的兴趣,也引发了各国政府和公众的关注。
(1)沉积物。主要是由上层海水中的颗粒物沉降到海底并不断堆积形成, 覆盖了近乎整个海底, 厚度从新形成洋壳上部几厘米到大陆边缘和深海沟的几千米不等[10]。海洋沉积物中的化学反应和运输主要通过扩散进行, 当然, 在一些流体活跃的位点如甲烷渗漏区和泥火山也存在物质的平流运输。深海沉积物根据与板块和大陆边缘的距离可以被大致分为主动大陆边缘、被动大陆架、大陆坡以及深海平原[11]。关于深部生物圈的有限认识目前主要来自于海洋沉积物,根据少量且集中在有机质丰富的大陆架沉积物中的生物量数据,Whitman等[12]估算认为埋藏在沉积物中的深部生物圈可能是地球上潜在的最大的生态系统,包含了全球1/10 ~1/3的生物量以及2/3的微生物[12]。并且,沉积物中的深部生物圈是一个埋藏了5.6×1010~3.03×1011 t C的巨大碳库,相当于全球植被的储碳量,同时也是一个巨大的营养物质储库[12]。
(2) 洋壳。洋壳中的岩石体积约是海洋沉积物总体积的5倍[11]。在全球大洋中,洋中脊广泛分布,总长度达6×104 km。炽热的岩浆沿着洋中脊不断涌出,与岩石圈相互作用,形成新的洋壳。海底每年形成的新洋壳约21 km3, 老的洋壳俯冲入板块边界消亡,平均6 100万年洋壳完成一次再循环[13,14]。洋壳主要由基性、超基性岩构成,含有丰富的铁、硫、镁等矿物。洋壳中流体的体积占到全球海水的2%,是地球上最大的含水系统[9]。玄武岩是顶层洋壳的主要岩石组分, 含有丰富的还原性的铁、锰和硫化物矿物(例如,二价铁约占9%, 二价锰和硫化物各约占0.1%),为微生物提供了相当可观的能量和营养源。同时,顶层500 m左右的洋壳岩石多孔隙且渗透性强[15],是微生物潜在的栖息地。流经洋壳的流体(来源于周围的底层海水)也可以为微生物带来氧气、硝酸盐和微量溶解有机碳(Dissolved Organic Carbon,DOC)等[16]。
(3) 热液喷口。在全球洋底分布着6×105 km2的洋中脊,大量在洋壳中被加热的流体沿着洋中脊不断渗出[9,17,18]。这些高温液体与岩浆岩相互作用,形成与周围海水化学性质迥异的高还原度且富含金属离子的液体。这些热液的化学组成变化不一,高度还原的热液与周围低温海水之间的不平衡导致矿物沉淀下来,形成热液烟囱和堆积体。热液口喷出的液体和随之被低温海水冷却形成的金属矿床是微生物的理想生境。反应产生的大量不稳定的还原性物质可作为微生物生长的能量来源, 是化能合成驱动的热液口生态系统的基础[19]。化学物质的多样性和动态变化导致了古菌和细菌的高度多样性: 不同热液口之间微生物群体组成不同, 同一个热液口随着化学和温度梯度微生物分布也不同,热液口的不同发育阶段也存在不同的微生物[20,21,22]。最近的研究结果表明, 热液口微生物的群落组成主要由热液流体的化学组成决定,其中H2浓度的影响尤为突出[23]。这些由热液驱动的水岩反应是全球地球化学循环的基本组成部分,对于理解洋壳和海水之间的物质循环和循环通量起到关键作用。
(4) 海底冷泉。冷泉生态系统是1979年首次发现于加利福尼亚州Borderland的圣克莱门特断裂带[24]。1983年在佛罗里达州的Escarpment也发现相似的生态系统,并被确认证实为冷泉生态系统[25]。冷泉是由深部沉积物中甲烷或者其他有机质流体向海底渗漏或者喷发而形成的独特环境[26]。由于冷泉流体的主要成分是甲烷,冷泉也被称为甲烷渗漏,冷泉流体的温度接近周围海水的温度。冷泉是由不同的地质活动,例如,板块潜没(Plate Subduction)、底辟作用(Diapirism)、重力压缩(Gravity Compression)或者水合物的分解所形成的。冷泉主要分布在不同地质板块交界的大陆架边缘,其渗漏甲烷的主要来源是深部沉积物中水合物的分解。全球的冷泉大部分分布于太平洋板块周围,是海底常见的生态系统。冷泉向上渗漏释放的有机质能够为海底大陆架边缘沉积物中的化能合成生态系统提供物质和能量。大陆架边缘的沉积物的地质物理化学环境(不同的物理化学环境包括温度、盐度、pH、氧气、二氧化碳、硫化氢、铵盐以及其他的无机挥发物质和金属物质等)变化范围大,限制了化能合成生物的生长。冷泉周围地化环境因子变化梯度连续且变化温和,为生活在其周围的化能合成生态系统中各种生物提供了适宜的栖息地[27]。与热液口类似,冷泉区形成了一个大型的生物量远高于周边海域的中心,而且多样性不高但生物量巨大。
研究深部环境中的生命无论是技术手段上还是分析方法上都是地球生命科学中最具挑战性的[7,17,28]。其中,研究样品必须要依靠科研钻探船才能获得,而现阶段只能通过参加全球大洋钻探计划组织的考察航次。历史上第一次致力于探究沉积物深部生物圈的大洋钻探航次为大洋钻探计划(Ocean Drilling Program,ODP)201航次[28],这次航次的成功让探索深部生物圈和深部海洋(subsurface ocean)成为了综合大洋钻探计划(Intergrated Ocean Drilling Program,IODP)中的一个核心主题。现在微生物学家们被邀请参加IODP航次已经成为常态,从2010年到目前为止已经执行完成了6个聚焦于深部生物圈的IODP航次,包括研究冲绳海槽沉积物中深热生物圈的331航次[29],研究南太平洋环流区极贫养(寡营养)沉积物中生命的329航次[30],研究大西洋中脊侧翼洋壳中深部生物圈的336航次[31],研究日本下北半岛附近海域深部煤层生物圈的337航次[32],研究大西洋中脊西侧翼亚特兰蒂斯蛇纹岩化与微生物活动的357航次[33],研究日本南海海槽深部生物圈温度极限的370航次[34]。预期基于这几个航次展开的数据分析和实验工作将给我们带来关于深部生物圈一些全新的甚至是颠覆性的认知。
深海平原约占全球海底面积的80%,通常分布于深度大于4 000 m的寡营养开放大洋。然而,对于深海平原沉积物的研究却非常欠缺。在寡营养大洋环流区开展的航次:南太平洋环流区的IODP329航次和北大西洋的IODP336航次,都旨在为开放大洋区域深部生物圈的组成和活性提供宝贵的信息[30,31]。其中,329航次主要以采集沉积物为主,研究碳源极度缺乏的沉积物中的生物圈。336航次则更为关注海床之下洋壳中的生物圈,并且建立了长期观测的实验装置[31]。开展的IODP331航次(2010年9月1日至10月4日)钻入了冲绳海槽的Iheya North热液区,旨在研究代谢方式多变的海底微生物生态系统以及周围的物理、化学特征。然而,这次航次并没能成功获取证明热液之下存在深热生物圈的直接证据,这需要未来更多的航次来实现。
(1) IODP357 Atlantis Massif (2015年10月26日至12月11日)
IODP357航次在大西洋中脊亚特兰蒂斯 “迷失之城”热液区(Lost City Hydrothermal Field, LCHF)完成了9个站位共17个钻孔的工作,钻孔深度为50~80 m。“迷失之城”热液系统极为特殊,之前在深海环境中从未发现过。其碳酸盐烟囱体中喷发出的流体高温(温度高达91 ℃),高pH值(9~11),缺乏金属离子和二氧化碳,但富含氢气、甲烷、有机酸和部分小分子化合物。其中,甲酸盐和小分子碳氢化合物可能由蛇纹岩化作用产生。IODP357航次的主要目的在于调查深部生物圈的活性、多样性,以及蛇纹岩化过程在维持地下微生物群落、促进热液系统的固碳过程中扮演的角色[33]。
(2) IODP 370 Muroto T-Limit (2016年9月10日至11月23日)
IODP370航次由日本“地球号”钻探船执行,采样点位于日本的南海海槽,由于较高的陆源沉积速率使得该处沉积层较厚。这次航次完成1个站位(C0023),获得深1 180 m的沉积物。在该站位,沉积物温度随深度增加。由于该区域的沉积物—基底界面处有非常活跃的热流活动,导致底部沉积物温度达到110~130 ℃,超出了已知的生命温度上界(120 ℃)。这让C0023成为探索深部生物圈温度极限的理想站位。航次旨在探究深部生物圈与温度的关系,详细研究控制微生物群落生物量、细胞活性和多样性的影响因子,以及通过检测沉积物的地球化学、地球物理以及水文参数来揭示深部生物圈的能量来源[34]。航次共获得112根岩芯柱,13 000份样品,其中部分样品直接由直升机转运到日本高知岩芯库,并在超净环境下完成分样和分析。由于在生命—非生命转化区(生命边界)微生物含量极低,所以这个航次对于污染的控制非常严格。获取的所有岩芯都会进行X射线扫描,并对用于微生物和地球化学分析的样品进行质量检测。对于质量较好的沉积物柱开展大量的地球化学、地质、沉积学和物理特性的检测,这个数据集对于定量分析深部生物的适应性起着重要的作用。航次在进行了原位温度测量的同时在钻孔中放置了13个热敏电阻,深度达863 m,用于长期监控原位温度。航次后的研究将着重分析尤其是生命边界区控制细胞大小、活性、多样性的环境因素,并确定限制和刺激微生物群落大小的理化和地质因素[34]。
(1) 发现洋壳生物圈
洋壳占据地球表面约65%的区域,体积是全球海洋沉积物的5倍[11]。然而受到采样装备和技术的限制,人们对洋壳的认知大多仅集中在地质学范畴,近年来的研究首先证明了高温(65 ℃)、缺氧的洋壳和裸露在海水中的玄武岩中存在微生物[16,35~37],进而发现更具有代表性的低温(<25 ℃)、有氧的洋壳环境中也存在微生物,其中的微生物对全球生物地球化学元素循环和海底地貌风化都具有潜在的重要作用[11,38~40]。分子系统发育学研究发现, 洋壳中的微生物群落具有高度的多样性, 其中的优势菌群有变形菌门(尤其是Alpha-和Gamma-proteobacteria),放线菌门(Actinobacteria),拟杆菌门(Bacteroidetes),绿弯菌门(Chloroflexi),厚壁菌门(Firmicutes)和浮霉菌门(Planctomycetes)[11,35,41]。通过调查位于大西洋洋中脊西侧翼的“North Pond”被沉积物覆盖洋壳中微生物的丰度、多样性和代谢潜力,发现每立方厘米的玄武岩中存在约104个细胞,细菌群落结构以与铁氧化密切相关的γ-变形菌和鞘脂杆菌为主。由于样品位点和数目的限制,目前对洋壳生物圈的规模尚无法准确估算。
(2) 沉积物生物圈规模的确定
海洋沉积物中蕴藏着丰富的微生物,我们对于深部生物圈现有的认知大多来自于海洋沉积物,因为通过大洋钻探计划可以较为容易地获取样品。早期推测沉积物中大约包含了超过1030个微生物,埋藏了5.6×1010~3.03×1011 t C,也是一个巨大的营养物质储库[12]。通过更有代表性的取样(扩展到寡营养大洋盆地)和更加准确的细胞计数手段,2012年Kallmeyer等[42]对沉积物生物圈的规模进行了更新,指出海洋沉积物中大约含有2.9×1029个微生物细胞,相当于4.1×1015g C,占地球总活体生物量的0.6%[42]。沉积物中绝大部分微生物种类不同于水体中的微生物,沉积物中的微生物细胞丰度与该区域的沉积速率和离岸距离有关,沉积速率越大,离岸距离越近,沉积物中微生物细胞数量越多。随着沉积物深度的增加,微生物细胞的丰度逐渐减少[42]。对于沉积物中微生物细胞以细菌还是古菌为主一直存在争议,最新的推测认为它们数量相当[43]。除了细菌和古菌,最近的研究认为真菌也是深部沉积物生物圈的重要组成部分,不可忽视,另外病毒也被认为在深部生物圈生态系统的演化中起到重要作用,预期这些方向的研究有可能在不久的将来会有突破。在超寡营养的南太平洋环流区海洋沉积物中,有氧呼吸的微生物群落可达海床下75 m,好氧菌可占据太平洋沉积物柱的15%~44%,以及全球海床的9%~37%[44]。目前探测到的埋藏最深的沉积物微生物来自于海底2.5 km以下。“地球号”IODP337航次获取了目前大洋钻探最深的沉积物样品,航次后期的研究工作发现在1.5~2.5 km深处的沉积物中存在煤层,微生物在40~60 ℃的环境下生活。甲烷和二氧化碳气体的同位素组成、生物标志物以及培养数据均指示微生物成因的甲烷。在1.5 km以下的沉积物中,微生物细胞丰度最高的为煤层区,其微生物群落与浅层的群落有显著差别,反而与有机质丰富的森林土壤中的群落更为相似,指示了陆源沉积物在沉积了上千万年后依然保留了原本的微生物群落且具有活性[45]。同时在深部沉积物中,以孢子形态生存于大约20 Ma BP的土壤中的69种进化上有差异的真菌被成功培养[46]。
(3) 检测确定深部细胞具有活性
2012年Lomstein等[47]率先利用D: L型氨基酸模型来定量微生物细胞、活的生物细胞周转时间、内生孢子和坏死细胞的生物量,首次指出沉积物中存在大量孢子,孢子数量与微生物细胞的数量相当;沉积物中微生物细胞的活性异常低,细胞生物量周转时间(microbial biomass turnover time)长达百年至千年。Lonstein领导的小组继续优化D: L型氨基酸模型,发展了利用坏死细胞中D: L-天门冬氨酸的消旋度构建模型推导微生物细胞活性,利用该模型推导出沉积物中的坏死细胞的周转时间平均大约几千年,微生物细胞的平均周转时间只有几年到几十年,比以前推测的时间缩短了100倍,因此在地质时间尺度上微生物的活性会对沉积物中的元素循环产生显著影响[48]。利用13C标记底物实验室富集培养检测,确定沉积物深层的微生物细胞可以吸收代谢标记底物,具有活性和功能[49,50]。
(4) 发现特殊代谢形式
深部生物圈中,大部分微生物类群没有亲缘关系较近的可培养菌株,因此长期以来我们对于这些类群的生理和代谢功能等的了解几乎是一片空白,其中的典型代表如深古菌门(Bathyarchaeota)和洛基古菌门(Lokiarchaeota)古菌。“深古菌” (Bathyarchaeota,曾经长期暂命名为“Miscellaneous Crenarchaeota Group”,MCG),是自然界分布非常广泛的一大类未培养古菌,在海洋沉积物中含量最为丰富,并且也是最活跃的类群之一。据推算“深古菌”在自然界的含量为2×1028 ~3.9×1028 个细胞,是地球上含量最丰富的微生物之一,很可能在全球物质和能量循环过程中发挥了重要的作用。单细胞基因组和元基因组分析发现了深古菌的代谢多样性和特殊性:其同时具有降解难降解有机化合物如芳香烃化合物、几丁质、纤维素等的代谢途径和利用无机碳自养合成乙酸的途径,是海洋沉积物中碳循环和生态系统的核心驱动者之一(图1)。He等[51]第一次发现和通过酶学功能检测证实古菌具有自养产乙酸的代谢方式,更新和极大地拓展了对古菌代谢多样性的认识,促使重新思考和理解古菌在深部生物圈的生态地位和功能,同时也为认识早期生命的起源打开了一扇新的窗户。同时,深古菌还可能具有甲烷氧化的功能,拓展了甲烷代谢微生物的类群[52]。
图1 海洋沉积物中深古菌介导的碳循环示意图
深古菌的一些类群具有丰富多样的代谢方式,既可以降解环境中的一些难降解的大分子如芳香化合物、几丁质、纤维素和蛋白质等,发酵产生乙酸等小分子化合物,也可以利用二氧化碳和H2自养产乙酸途径获取能量。深古菌产生的乙酸是重要的电子载体,可以被甲烷产生菌和异养细菌所利用,是深部碳循环和生态系统的重要驱动者
Fig.1 Roles of Bathyarchaeota in carbon cycling in marine sediments
Bathyarchaeota contains diversified subgroups and possesses versitile metabolic pathways. They can degrade different refractory macromolecules, such as aromatic compounds, cellulose, chitin, and extracellular protein, into small molucules such as acetate by fermentation in sedimentary environments. Meanwhile, they can obtain energy from autotrophic pathway by producing acetate from hydrogen and carbon dioxide.
The acetate produced by Bathyarchaeota is an important electron accepter that can be utilized by methanogens and heterotrophic bacteria, which makes Bathyarcaeota an important driver of deep carbon cycle and ecosystems
如上介绍,通过几十年来科学家不懈的探索,目前科学界已普遍认识到海底隐藏着一个巨大的深部生物圈,对深部生物圈的研究也成为大洋钻探2013—2023年研究规划的四大研究主题之一。最重要和突出的研究进展包括:确定了沉积物中生物圈的规模,微生物细胞数量与海水细胞数量相当;微生物细胞代谢缓慢但仍然具有活性,其平均代增时间为几年到几十年;确定了沉积物中微生物的类型,发现了与微生物细胞数量相当的孢子,发现了沉积物中一些重要古菌在生命演化和元素循环中发挥了重要作用;认识并验证了深部生物圈的一个重要能量来源和支撑为H2,并且乙酸也是深部生态系统重要的物质能量来源;发现了洋壳生态系统等。然而对于这个庞大的隐藏生态系统,有更多的未知科学问题等待探索和发现。当前深部生物圈研究的前沿核心科学问题包括:①决定深部生命分布、生命—非生命边界转换的主要环境因子是什么?是什么机制?②深部生命的生物地球化学功能和机制是什么?对地球元素循环的贡献多少?③深部生命生存代谢、适应和演化的机制是什么?病毒是否是推动深部生态系统演化的主要或关键因子?④深部生态系统与上层生态系统的相互联系与影响的内在关系是什么?具体的机制是什么?⑤地球环境变化是否/如何与深部生命相互影响?深部生命是否记载了古气候变化的历史?如何解读并帮助回望地球历史并预测未来?
回答这些前沿科学问题面临巨大的困难和挑战,例如,传统的海底科学调查方法是直接取样后在船上或岸上实验室开展实验分析,但不能反映生物的原位功能和活性;深部生命面临极端的环境条件很难在实验室模拟;深部生命代谢缓慢如何在实验室培养开展研究?深部无污染的岩石样品获取非常困难等。近年来,科学技术的快速发展为海底调查带来了原位观测系统以及一系列先进的工具和技术,例如,海底井控观测装置(Circulation Obviation Retrofit Kit,CORK)[53],为多学科进行水下考察带来了前所未有的机遇,也让我们能够更加全面地了解深海微生物活动与海底水文条件和海水化学参数之间的关系[54,55,56]。除了海底原位实验之外,先进的实验室生化反应器可以仿照自然界海水环境并用于模拟微生物的生长[57,58,59,60]。高压是模拟深海或是海底环境时最为关键的环境因素之一,根据研究目的和所用样品的来源,不同类型的高压反应器(高液压和高气压生物反应器)被用于模拟原位环境,高压生物反应器能极大地帮助深入研究深部生物圈,例如,Zhang等[61]设计了高压流动培养装置用于富集沉积物中的厌氧甲烷氧化菌。
对于深部生物圈的研究需要摆脱基于分类学以描述为主的阶段,开展多学科的定量原位实验,需要与微生物、水文、地球化学和地球物理研究结合。新的分子技术如稳定同位素探针技术(SIP)、单细胞分离测序技术[62]、荧光原位杂交—二次离子色谱(FISH-SIMS)[63,64]、下一代组学OMICs技术(宏基因组、宏转录组、宏蛋白质组和宏代谢组)[65,66,67]的运用, 传统和现代手段的相互结合, 将会为我们深入研究深部微生物多样性、起源和演化, 生理活性和地球化学、生态学功能提供前所未有的机会。同时要将小范围尺度的深部微生物学研究与大范围尺度的生物地球化学过程耦合具有很大的挑战性,其中一个解决方法是建立将水文、地球化学、地球物理和微生物学这些多学科的研究结合起来的原位观测平台。该平台的建立将使深部生物实验从岸上的实验室转移到原位的深海环境,从研究个别微生物类群发展到定量观测深部沉积物中整个微生物群落。
中国在深部生物圈方向的研究才刚刚起步,而且限于采样技术手段的限制目前尚没有中国科学家主导的深部生物圈研究ODP/IODP航次,只是通过派出微生物学家参加大洋钻探航次,培养开展深部生物圈研究的科学家取得了一些成绩。自2010年至今,中国ODP/IODP共派出6位微生物科学家参加了7个大洋钻探航次。来自南京大学的刘常宏通过参加IODP337航次获得无污染的深部煤层沉积物样品开展真菌分离和鉴定的工作,确定了深部沉积物中确实存在可培养的真菌,从深部煤层沉积物中成功分离培养了69种进化上有差异的真菌,产生了较好的国际影响[46]。中国海洋大学张晓华参加IODP航次从寡营养的深海沉积物中分离了大量不同的细菌新种[68]。上海交通大学王风平研究团队首次证明了年轻、低温、有氧的北大西洋中脊(North Pond)玄武岩洋壳中含有独特且活跃的微生物类群,这些微生物很可能参与了与铁元素相关的洋壳风化作用[39]。此外,还揭示了外源氮源的添加可以刺激洋壳微生物的生长的现象,暗示了氮源是洋壳生物圈中微生物生长的潜在限制因子之一[40]。厦门大学张瑶等[69]作为IODP航次的岸上科学家研究了南海长达800 m的沉积物柱中细菌群落结构与地质过程、环境参数等影响因子的关系,发现相比于环境参数和沉积年代,不同层位之间的地理隔离对塑造细菌群落结构起更加重要的作用。同时也发现沉积物深部的微生物可能靠分解沉积物中的有机质维持生命,并改变了沉积物中有机质的含量和类型。上海海洋大学方家松对IODP337航次获得的煤层沉积物进行实验室高压模拟培养,发现部分嗜压细菌在20 Ma BP前的煤层沉积物中以孢子的形态存在[70]。除了通过参加IODP航次开展深部生物圈研究,随着深海载人和无人深潜器工程技术的发展和应用,近年来中国科学家在深海热液口和冷泉区开展了大量的工作。例如,香港科技大学的钱培元团队运用宏基因组的方法研究深海热液区微生物参与的硫元素循环[71]。国家海洋局第三海洋研究所邵宗泽团队对热液口极端环境中的细菌、古菌进行分离培养[72,73,74]。上海交通大学肖湘研究团队从深海热液口分离了嗜热嗜高压古菌[75,76],并提出了古菌极端条件适应的核心共同适应机制[77,78]。上海交通大学王风平研究团队对南海冷泉区沉积物中甲烷代谢古菌的分布、多样性和代谢特征进行了研究[79,80]等。
但是中国开展深部生物圈研究总体上还相对零散,缺乏复合型人才和整体技术特色/优势,因此中国发展深部生物圈研究需要有更多的顶层设计,凝练聚焦关键科学问题协同攻关。在近期,借助作为IODP完全成员国(full member)的身份依托参加IODP航次的机会培养一批致力于深部生物圈研究的优秀青年中国科学家,鼓励中国科学家参与和牵头相关国际研究计划的讨论、设计和组织,形成一些特色技术和优势方向。进而,以深部生物圈研究为核心,围绕深部碳循环、深部生命极限和深部生命的适应、演化三大主题,综合考虑和整合中国地球科学的优势学科,开展包括多个不同学科(如工程技术、地球化学、地质构造、分子生物学等)高度交叉融合的多学科研究,建设高水平深部生物圈研究平台和中心,培育和促进多学科的交叉和交融,实现引领国际深部碳循环和深部生命科学研究的长远目标。
致 谢:感谢贾泽宇同学在文章撰写过程中提供的帮助。
The authors have declared that no competing interests exist.
| [1] |
Feast and famine—Microbial life in the deep-sea bed [J].
The seabed is a diverse environment that ranges from the desert-like deep seafloor to the rich oases that are present at seeps, vents, and food falls such as whales, wood or kelp. As well as the sedimentation of organic material from above, geological processes transport chemical energy--hydrogen, methane, hydrogen sulphide and iron--to the seafloor from the subsurface below, which provides a significant proportion of the deep-sea energy. At the sites on the seafloor where chemical energy is delivered, rich and diverse microbial communities thrive. However, most subsurface microorganisms live in conditions of extreme energy limitation, with mean generation times of up to thousands of years. Even in the most remote subsurface habitats, temperature rather than energy seems to set the ultimate limit for life, and in the deep biosphere, where energy is most depleted, life might even be based on the cleavage of water by natural radioisotopes. Here, we review microbial biodiversity and function in these intriguing environments.
|
| [2] |
Vertical distribution of bacteria in marine sediments [J].
Off the coast of Southern California bacteria are more abundant in marine sediments than in the overlying water, there being thousands to millions per gram of sediment and only a few hundred or less per milliliter of water. The distribution of bacteria in the sediment is more or less independent of the depth of the overlying water, the temperature of the ocean floor, and the distance from mainland. The bacterial population seems to be influenced more greatly by the organic content of the sediments than by other factors. Vertical sections from cores 40 to 75 cm. in length show that the number of bacteria in the sediments decreases greatly with depth in the upper 5 cm. of the core and more slowly beyond. In the surface slime, aerobes are 5 to 100 times more plentiful than a aerobes, but at depths exceeding 15 centimeters about as many anaerobes as aerobes are present. Both anaerobes and aerobes have been demonstrated at all depths analyzed. The oxidation-reduction potential (intensity factor expressed as E) increases with core depth, although the oxygen-absorbing capacity decreases with depth. Both properties are ascribed primarily to bacterial activity. Bacteria recovered from marine sediments exhibit many biochemical processes such as ammonification, proteolysis, nitrate reduction, nitrification, urea fermentation, chitin digestion, methane production, fat hydrolysis, cellulose decomposition, glucose, xylose, and arabinose fermentation and starch hydrolysis.
|
| [3] |
Barophilic bacteria in some deep sea sediments [J].
ZOBELL CE, MORITA RY.
|
| [4] |
Studies on the bacterial flora of marine bottom sediments [J].
The bacteriological analysis of 126 sediment samples collected in the Channel Island region along the coast of Southern California from bottoms as deep as 2,000 meters reveals the presence of several physiological types of bacteria which may influence the diagenesis of recent sediments. Strict aerobes recovered from the bottoms of cores over 50 cm long where there is no free oxygen may have been buried in the sediments in a dormant state for a long time. Core depth, organic-matter content, and the median particle size of the sediments are the chief factors which influence the bacterial populations. Most of the bacteria are capable of multiplication and biochemical activity at 0 degrees C. Bacteria which decompose proteins, cellulose, starch, chitin, and other complex organic compounds are quite abundant in the bottom deposits. The occurrence of lipoclastic species which utilize the glycerol from fats and leave long-chain fatty acids may help account for the genesis of petroleum.
|
| [5] |
Submarine thermal springs on the Galapagos rift [J]. |
| [6] |
Deep bacterial biosphere in Pacific Ocean sediments [J].
ALTHOUGH around 70% of the Earth's surface is marine, little is known about the microbiology of underlying sediments, which can be more than a kilometre deep 1 . Selective degradation of organic matter within sediments over geological time profoundly affects the chemical composition of the ocean and atmosphere 2 . Microbial processes have a fundamental role in surface sediments 3,4 , but despite geochemical evidence 5 , their significance in deeper sediments has not been established 6 . Here we report the discovery of viable sediment bacterial populations at five Pacific Ocean sites to depths >500m. Bacterial distributions and activities are commensurate with geochemical changes. Bacterial profiles with depth are remarkably consistent and deviations can be linked to specific environmental factors. The rate of decline in these populations indicates that bacteria are present to even greater depths.
|
| [7] |
Discovering the roles of subsurface microorganisms: Progress and future of deep biosphere investigation [J]. |
| [8] |
Microbial provinces in the subseafloor [J].
Abstract The rocks and sediments of the oceanic subsurface represent a diverse mosaic of environments potentially inhabited by microorganisms. Understanding microbial ecosystems in subseafloor environments confounds standard ecological descriptions in part because we have difficulty elucidating and describing the scale of relevant processes. Habitat characteristics impact microbial activities and growth, which in turn affect microbial diversity, net production, and global biogeochemical cycles. Herein we provide descriptions of subseafloor microbial provinces, broadly defined as geologically and geographically coherent regions of the subseafloor that may serve as potential microbial habitats. The purpose of this review is to summarize and refine criteria for the definition and delineation of distinct subseafloor microbial habitats to aid in their exploration. This review and the criteria we outline aim to develop a unified framework to improve our understanding of subseafloor microbial ecology, enable quantification of geomicrobial processes, and facilitate their accurate assimilation into biogeochemical models.
|
| [9] |
Fluxes of fluid and heat from the oceanic crustal reservoir [J].
Recent discoveries define a global scale fluid reservoir residing within the uppermost igneous oceanic crust, a region of seafloor that is both warm and may harbor a substantial biosphere. This hydrothermal fluid reservoir formed initially within volcanic rocks newly erupted at mid-ocean ridges, but extends to the vastly larger and older ridge flanks. Upper oceanic crust is porous and permeable due to the presence of lava drainbacks, fissuring, and inter-unit voids, and this porosity and permeability allows active fluid circulation to advect measurable quantities of lithospheric heat from the crust to an average age of 65 Myr. A compilation of crustal porosities shows that this fluid reservoir contains nearly 2% of the total volume of global seawater. Heat flow and sediment thickness data allow calculation of reservoir temperatures, predicting 40掳C mean temperatures in Cretaceous crust. Utilizing these temperature estimates, heat flow measurements and models for the thermal structure and evolution of the oceanic lithosphere, we have computed mean hydrothermal fluxes into the deep ocean as a function of plate age. The total hydrothermal volume flux into the oceans approaches 20% of the total riverine input and may contribute to the global seawater mass balance.
|
| [10] |
Prokaryotic biodiversity and activity in the deep subseafloor biosphere [J].
Abstract Top of page Abstract Introduction Prokaryotic biodiversity Prokaryotic activity Prospects for the future Acknowledgements References The deep subseafloor biosphere supports a diverse population of prokaryotes belonging to the Bacteria and Archaea . Most of the taxonomic groups identified by molecular methods contain mainly uncultured phylotypes. Despite this several cultured strains have been isolated from this habitat, but they probably do not represent the majority of the population. Evidence is starting to suggest that some of the activities measured, such as sulphate reduction and methanogenesis, reflected in geochemical profiles, are carried out by a small subset of the community detected by molecular methods. It is further possible that heterotrophy may be the most important mode of metabolism in subsurface sediments and heterotrophic microorganisms could dominate the uncultured prokaryotic population. Although, heterotrophy is limited by the increasing recalcitrance of organic matter with depth, this may be counteracted by thermal activation of buried organic matter providing additional substrates at depth.
|
| [11] |
Microbial ecology of the dark ocean above, at, and below the seafloor [J].
The majority of life on Earth--notably, microbial life--occurs in places that do not receive sunlight, with the habitats of the oceans being the largest of these reservoirs. Sunlight penetrates only a few tens to hundreds of meters into the ocean, resulting in large-scale microbial ecosystems that function in the dark. Our knowledge of microbial processes in the dark ocean-the aphotic pelagic ocean, sediments, oceanic crust, hydrothermal vents, etc.-has increased substantially in recent decades. Studies that try to decipher the activity of microorganisms in the dark ocean, where we cannot easily observe them, are yielding paradigm-shifting discoveries that are fundamentally changing our understanding of the role of the dark ocean in the global Earth system and its biogeochemical cycles. New generations of researchers and experimental tools have emerged, in the last decade in particular, owing to dedicated research programs to explore the dark ocean biosphere. This review focuses on our current understanding of microbiology in the dark ocean, outlining salient features of various habitats and discussing known and still unexplored types of microbial metabolism and their consequences in global biogeochemical cycling. We also focus on patterns of microbial diversity in the dark ocean and on processes and communities that are characteristic of the different habitats.
|
| [12] |
Prokaryotes: The unseen majority [J]. |
| [13] |
Heat flow from the Earth’s interior: Analysis of the global data set [J]. |
| [14] |
Seafloor spreading: Portrait of a magma chamber [J].
Focuses on seafloor spreading and the molten rock (melt or magma) that solidifies into ocean crust or attaches to the edges of the separating tectonic plates. Reference to a study in this issue by Kent and co-workers, which produced the first three-dimensional seismic reflection images of a mid-ocean-ridge magma system; Description of two models that were proposed to explain how melt is supplied to the crust along fast-opening spreading centers such as the East Pacific Rise.
|
| [15] |
Marine hydrogeology: Recent accomplishments and future opportunities [J].
Marine hydrogeology is a broad-ranging scientific discipline involving the exploration of fluid-搑ock interactions below the seafloor. Studies have been conducted at seafloor spreading centers, mid-plate locations, and in plate- and continental-margin environments. Although many seafloor locations are remote, there are aspects of marine systems that make them uniquely suited for hydrologic analysis. Newly developed tools and techniques, and the establishment of several multidisciplinary programs for oceanographic exploration, have helped to push marine hydrogeology forward over the last several decades. Most marine hydrogeologic work has focused on measurement or estimation of hydrogeologic properties within the shallow subsurface, but additional work has emphasized measurements of local and global fluxes, fluid source and sink terms, and quantitative links between hydrogeologic, chemical, tectonic, biological, and geophysical processes. In addition to summarizing selected results from a small number of case studies, this paper includes a description of several new experiments and programs that will provide outstanding opportunities to address fundamental hydrogeologic questions within the seafloor during the next 20-30 years.
|
| [16] |
Inorganic chemistry, gas compositions and dissolved organic carbon in fluids from sedimented young basaltic crust on the Juan de Fuca Ridge flanks [J].
The permeable upper oceanic basement serves as a plausible habitat for a variety of microbial communities. There is growing evidence suggesting a substantial subseafloor biosphere. Here new time series data are presented on key inorganic species, methane, hydrogen and dissolved organic carbon (DOC) in ridge flank fluids obtained from subseafloor observatory CORKs (Circulation Obviation Retrofit Kits) at Integrated Ocean Drilling Program (IODP) boreholes 1301A and 1026B. These data show that the new sampling methods (Cowen et al., 2012) employed at 1301A result in lower contamination than earlier studies. Furthermore, sample collection methods permitted most chemical analyses to be performed from aliquots of single large volume samples, thereby allowing more direct comparison of the data. The low phosphate concentrations (0.06–0.2μM) suggest that relative to carbon and nitrogen, phosphorus could be a limiting nutrient in the basement biosphere. Coexisting sulfate (17–18mM), hydrogen sulfide (650.1μM), hydrogen (0.3–0.7μM) and methane (1.5–2μM) indicates that the basement aquifer at 1301A either draws fluids from multiple flow paths with different redox histories or is a complex environment that is not thermodynamically controlled and may allow co-occurring metabolic pathways including sulfate reduction and methanogenesis. The low DOC concentrations (11–18μM) confirm that ridge flank basement is a net DOC sink and ultimately a net carbon sink. Based on the net amounts of DOC, oxygen, nitrate and sulfate removed (6530μM, 6580μM, 6540μM and 6510mM, respectively) from entrained bottom seawater, organic carbon may be aerobically or anaerobically oxidized in biotic and/or abiotic processes.
|
| [17] |
McCollom T M. Geomicrobiology in oceanography: Microbe-mineral interactions at and below the seafloor [J].
Abstract Oceanography is inherently interdisciplinary and, since its inception, has included the study of microbe-mineral interactions. From early studies of manganese nodules, to the discovery of hydrothermal vents, it has been recognized that microorganisms are involved at various levels in the transformation of rocks and minerals at and below the seafloor. Recent studies include mineral weathering at low temperatures and microbe-mineral interactions in the subseafloor "deep biosphere". A common characteristic of seafloor and subseafloor geomicrobiological processes that distinguishes them from terrestrial or near-surface processes is that they occur in the dark, one or more steps removed from the sunlight that fuels the near-surface biosphere on Earth. This review focuses on geomicrobiological studies and energy flow in dark, deep-ocean and subseafloor rock habitats.
|
| [18] |
Oceanic phosphorus imbalance: Magnitude of the mid-ocean ridge flank hydrothermal sink [J].
We present a new estimate for the crustal phosphorous sink that results from reactions among seawater, basalt, and sediment blanketing low temperature mid-ocean ridge flank hydrothermal systems. New estimates for global hydrothermal power output, sediment thickness, and the dissolved phosphate concentrations in basement formation fluids indicate that fluid flow through ridge flanks removes 2.8 脳 10mol P yr. This value is larger (130%) than the riverine dissolved flux of inorganic phosphate and is as much as 35% of the sedimentary P sink. The concordant seawater flux (2.1 脳 10kg yr) is 65% of the riverine fluid flux and circulates a fluid volume equivalent to the entire ocean in about 70,000 yr. Additional sampling of seafloor springs is required to further constrain the range of calculated phosphate fluxes; nevertheless the modern phosphorus budget is clearly unbalanced with total sinks outpacing sources.
|
| [19] |
Biogeochemistry: Microbial essentials at hydrothermal vents [J].
Nature is the international weekly journal of science: a magazine style journal that publishes full-length research papers in all disciplines of science, as well as News and Views, reviews, news, features, commentaries, web focuses and more, covering all branches of science and how science impacts upon all aspects of society and life.
|
| [20] |
GeoChip-based analysis of metabolic diversity of microbial communities at the Juan de Fuca Ridge hydrothermal vent [J]. |
| [21] |
Temporal changes in archaeal diversity and chemistry in a mid-ocean ridge subseafloor habitat [J]. |
| [22] |
Temporal and spatial archaeal colonization of hydrothermal vent deposits [J].
Summary Thermocouple arrays were deployed on two deep-sea hydrothermal vents at Guaymas Basin (27°0.5′N, 111°24.5′W) in order to measure in situ temperatures at which microorganisms colonize the associated mineral deposits. Intact sections of three structures that formed around the arrays were collected after 4 and 72day deployments (named BM4, BM72 and TS72). Archaeal diversity associated with discreet subsamples collected across each deposit was determined by polymerase chain reaction amplification of 16S rRNA genes. Spatial differences in archaeal diversity were observed in all deposits and appeared related to in situ temperature. In BM4, no 16S rRNA genes were detected beyond about 1.5cm within the sample (>200°C). Phylotypes detected on the outside of this deposit belong to taxonomic groups containing mesophiles and (hyper)thermophiles, whereas only putative hyperthermophiles were detected 1.5cm inside the structure (65110°C). In contrast, the more moderate thermal gradient recorded across TS72 was associated with a deeper colonization (2–3cm inside the deposit) of putative hyperthermophilic phylotypes. Although our study does not provide a precise assessment of the highest temperature for the existence of microbial habitats inside the deposits, archaeal 16S rRNA genes were detected directly next to thermocouples that measured 110°C ( Methanocaldococcus spp. in BM4) and 116°C ( Desulfurococcaceae in TS72). The successive array deployments conducted at the Broken Mushroom (BM) site also revealed compositional differences in archaeal communities associated with immature (BM4) and mature chimneys (BM72) formed by the same fluids. These differences suggest a temporal transition in the primary carbon sources used by the archaeal communities, with potential CO 2 /H 2 methanogens prevalent in BM4 being replaced by possible methylotroph or acetoclastic methanogens and heterotrophs in BM72. This study is the first direct assessment of in situ conditions experienced by microorganisms inhabiting actively forming hydrothermal deposits at different stages of structure development.
|
| [23] |
Microbial community structure of hydrothermal deposits from geochemically different vent fields along the Mid-Atlantic Ridge [J].
Summary To evaluate the effects of local fluid geochemistry on microbial communities associated with active hydrothermal vent deposits, we examined the archaeal and bacterial communities of 12 samples collected from two very different vent fields: the basalt-hosted Lucky Strike (37°17′N, 32°16.3′W, depth 1600–1750m) and the ultramafic-hosted Rainbow (36°13′N, 33°54.1′W, depth 2270–2330m) vent fields along the Mid-Atlantic Ridge (MAR). Using multiplexed barcoded pyrosequencing of the variable region 4 (V4) of the 16S rRNA genes, we show statistically significant differences between the archaeal and bacterial communities associated with the different vent fields. Quantitative polymerase chain reaction (qPCR) assays of the functional gene diagnostic for methanogenesis ( mcr A), as well as geochemical modelling to predict pore fluid chemistries within the deposits, support the pyrosequencing observations. Collectively, these results show that the less reduced, hydrogen-poor fluids at Lucky Strike limit colonization by strict anaerobes such as methanogens, and allow for hyperthermophilic microaerophiles, like Aeropyrum . In contrast, the hydrogen-rich reducing vent fluids at the ultramafic-influenced Rainbow vent field support the prevalence of methanogens and other hydrogen-oxidizing thermophiles at this site. These results demonstrate that biogeographical patterns of hydrothermal vent microorganisms are shaped in part by large scale geological and geochemical processes.
|
| [24] |
A deep-sea hydrothermal site on a strike-slip fault [J].
Submersible exploration of a young scarp in the San Clemente fault zone, at 1,800 m in the California Borderland, discovered tall piles of hydrothermal barite and dense colonies of large benthic animals. Phenomena along this active strike-slip fault zone resemble those at hot springs along the axes of mid-ocean ridges.
|
| [25] |
Biological communities at the Florida Escarpment resemble hydrothermal vent taxa [J].
Dense biological communities of large epifaunal taxa similar to those found along ridge crest vents at the East Pacific Rise were discovered in the abyssal Gulf of Mexico. These assemblages occur on a passive continental margin at the base of the Florida Escarpment, the interface between the relatively impermeable hemipelagic clays of the distal Mississippi Fan and the jointed Cretaceous limestone of the Florida Platform. The fauna apparently is nourished by sulfide rich hypersaline waters seeping out at near ambient temperatures onto the sea floor.
|
| [26] |
Reproduction and dispersal at vents and cold seeps [J]. |
| [27] |
Ecology of cold seep sediments: Interactions of fauna with flow, chemistry and microbes [J].
Cold seeps occur in geologically active and passive continental margins, where pore waters enriched in methane are forced upward through the sediments by pressure gradients. The advective supply of methane leads to dense microbial communities with high metabolic rates. Anaerobic methane oxidation presumably coupled to sulphate reduction facilitates formation of carbonates and, in many places, generates extremely high concentrations of hydrogen sulphide in pore waters. Increased food supply, availability of hard substratum and high concentrations of methane and sulphide supplied to free-living and symbiotic bacteria provide the basis for the complex ecosystems found at these sites. This review examines the structures of animal communities in seep sediments and how they are shaped by hydrologic, geochemical and microbial processes. The full size range of biota is addressed but emphasis is on the mid-size sediment-dwelling infauna (foraminiferans, metazoan meiofauna and macrofauna), which have received less attention than megafauna or microbes. Megafaunal biomass at seeps, which far exceeds that of surrounding non-seep sediments, is dominated by bivalves (mytilids, vesicomyids, lucinids and thyasirids) and vestimentiferan tube worms, with pogonophorans, cladorhizid sponges, gastropods and shrimp sometimes abundant. In contrast, seep sediments at shelf and upper slope depths have infaunal densities that often differ very little from those in ambient sediments. At greater depths, seep infauna exhibit enhanced densities, modified composition and reduced diversity relative to background sediments. Dorvilleid, hesionid and ampharetid polychaetes, nematodes, and calcareous foraminiferans are dominant. There is extensive spatial heterogeneity of microbes and higher organisms at seeps. Specialized infaunal communities are associated with different seep habitats (microbial mats, clam beds, mussel beds and tube worms aggregations) and with different vertical zones in the sediment. Whereas fluid flow and associated porewater properties, in particular sulphide concentration, appear to regulate the distribution, physiological adaptations and sometimes behaviour of many seep biota, sometimes the reverse is true. Animal-microbe interactions at seeps are complex and involve symbioses, heterotrophic nutrition, geochemical feedbacks and habitat structure. Nutrition of seep fauna varies, with thiotrophic and methanotrophic symbiotic bacteria fueling most of the megafaunal forms but macrofauna and most meiofauna are mainly heterotrophic. Macrofaunal food sources are largely photosynthesis-based at shallower seeps but reflect carbon fixation by chemosynthesis and considerable incorporation of methane-derived C at deeper seeps. Export of seep carbon appears to be highly localized based on limited studies in the Gulf of Mexico. Seep ecosystems remain one of the ocean's true frontiers. Seep sediments represent some of the most extreme marine conditions and offer unbounded opportunities for discovery in the realms of animal-microbe-geochemical interactions, physiology, trophic ecology, biogeography, system-atics and evolution.
|
| [28] |
Proceedings of the Ocean Drilling Program, Initial Reports,201[R].Ocean Drilling Program, |
| [29] |
Deep Hot Biosphere[R]. Integrated Ocean Drilling Program Expedition 331 Preliminary Report. Washington DC:Integrated Ocean Drilling Program Management Internationalxpedition 331 Scientists. Deep Hot Biosphere[R]. Integrated Ocean Drilling Program Expedition 331 Preliminary Report . |
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
Expedition 357 Preliminary Report: Atlantis Massif Serpentinization and Life[R] . |
| [34] |
Expedition 370 preliminary report: Temperature limit of the deep biosphere off muroto [J].
International Ocean Discovery Program (IODP) Expedition 370 aimed to explore the limits of life in the deep subseafloor biosphere at a location where temperature increases with depth at an intermediate rate and exceeds the known temperature maximum of microbial life (~120°C) at the sediment/basement interface ~1.2 km below the seafloor. Drilling Site C0023 is located in the vicinity of Ocean Drilling Program (ODP) Sites 808 and 1174 at the protothrust zone in the Nankai Trough off Cape Muroto at a water depth of 4776 m. ODP Leg 190 in 2000, revealed the presence of microbial cells at Site 1174 to a depth of ~600 meters below seafloor (mbsf), which corresponds to an estimated temperature of ~70°C, and reliably identified a single zone of higher cell concentrations just above the décollement at around 800 mbsf, where temperature presumably reached 90°C; no cell count data was reported for other sediment layers in the 70°–120°C range, because the limit of manual cell count for low-biomass samples was not high enough. With the establishment of Site C0023, we aimed to detect and investigate the presence or absence of life and biological processes at the biotic–abiotic transition with unprecedented analytical sensitivity and precision. Expedition 370 was the first expedition dedicated to subseafloor microbiology that achieved time-critical processing and analyses of deep biosphere samples by simultaneous shipboard and shore-based investigations.Our primary objectives during Expedition 370 were to study the relationship between the deep subseafloor biosphere and temperature. We aimed to comprehensively study the factors that control biomass, activity, and diversity of microbial communities in a subseafloor environment where temperatures increase from ~2°C at the seafloor to ~120°C at the sediment/basement interface and thus likely encompasses the biotic–abiotic transition zone. We also aimed to determine geochemical, geophysical, and hydrogeological characteristics in sediment and the underlying basaltic basement and elucidate if the supply of fluids containing thermogenic and/or geogenic nutrient and energy substrates may support subseafloor microbial communities in the Nankai accretionary complex.To address these primary scientific objectives and questions, we penetrated 1180 m and recovered 112 cores across the sediment/basalt interface. More than 13,000 samples were collected, and selected samples were transferred to the Kochi Core Center by helicopter for simultaneous microbiological sampling and analysis in laboratories with a super-clean environment. Following the coring operations, a temperature observatory with 13 thermistor sensors was installed in the borehole to 863 mbsf.
|
| [35] |
Abundance and diversity of microbial life in ocean crust [J].
Oceanic lithosphere exposed at the sea floor undergoes seawater-rock alteration reactions involving the oxidation and hydration of glassy basalt. Basalt alteration reactions are theoretically capable of supplying sufficient energy for chemolithoautotrophic growth. Such reactions have been shown to generate microbial biomass in the laboratory, but field-based support for the existence of microbes that are supported by basalt alteration is lacking. Here, using quantitative polymerase chain reaction, in situ hybridization and microscopy, we demonstrate that prokaryotic cell abundances on seafloor-exposed basalts are 3-4 orders of magnitude greater than in overlying deep sea water. Phylogenetic analyses of basaltic lavas from the East Pacific Rise (9 degrees N) and around Hawaii reveal that the basalt-hosted biosphere harbours high bacterial community richness and that community membership is shared between these sites. We hypothesize that alteration reactions fuel chemolithoautotrophic microorganisms, which constitute a trophic base of the basalt habitat, with important implications for deep-sea carbon cycling and chemical exchange between basalt and sea water.
|
| [36] |
Fluids from aging ocean crust that support microbial life [J].
Abstract Little is known about the potential for life in the vast, low-temperature (<100 degrees C) reservoir of fluids within mid-ocean ridge flank and ocean basin crust. Recently, an overpressured 300-meter-deep borehole was fitted with an experimental seal (CORK) delivering crustal fluids to the sea floor for discrete and large-volume sampling and characterization. Results demonstrate that the 65 degrees C fluids from 3.5-million-year-old ocean crust support microbial growth. Ribosomal RNA gene sequence data indicate the presence of diverse Bacteria and Archaea, including gene clones of varying degrees of relatedness to known nitrate reducers (with ammonia production), thermophilic sulfate reducers, and thermophilic fermentative heterotrophs, all consistent with fluid chemistry.
|
| [37] |
Microbial diversity within basement fluids of the sediment-buried Juan de Fuca Ridge flank [J].
Despite its immense size, logistical and methodological constraints have largely limited microbiological investigations of the subseafloor basement biosphere. In this study, a unique sampling system was used to collect fluids from the subseafloor basaltic crust via a Circulation Obviation Retrofit Kit (CORK) observatory at Integrated Ocean Drilling Program borehole 1301A, located at a depth of 2667 m in the Pacific Ocean on the eastern flank of the Juan de Fuca Ridge. Here, a fluid delivery line directly accesses a 3.5 million years old basalt-hosted basement aquifer, overlaid by 262 m of sediment, which serves as a barrier to direct exchange with bottom seawater. At an average of 1.2 x 10(4) cells ml(-1), microorganisms in borehole fluids were nearly an order of magnitude less abundant than in surrounding bottom seawater. Ribosomal RNA genes were characterized from basement fluids, providing the first snapshots of microbial community structure using a high-integrity fluid delivery line. Interestingly, microbial communities retrieved from different CORKs (1026B and 1301A) nearly a decade apart shared major community members, consistent with hydrogeological connectivity. However, over three sampling years, the dominant gene clone lineage changed from relatives of Candidatus Desulforudis audaxviator within the bacterial phylum Firmicutes in 2008 to the Miscellaneous Crenarchaeotic Group in 2009 and a lineage within the JTB35 group of Gammaproteobacteria in 2010, and statistically significant variation in microbial community structure was observed. The enumeration of different phylogenetic groups of cells within borehole 1301A fluids supported our observation that the deep subsurface microbial community was temporally dynamic. The ISME Journal (2013) 7, 161-172; doi:10.1038/ismej.2012.73; published online 12 July 2012
|
| [38] |
Abundance, Diversity and Metabolic Potentials of Subsurface Crustal Microorganisms on the Western Flank of the Mid-Atlantic Ridge[D] .大西洋中脊西侧翼洋壳微生物多样性、功能和代谢潜能研究[D] . |
| [39] |
Diversity and metabolic potentials of subsurface crustal microorganisms from the western flank of the Mid-Atlantic Ridge [J].
Deep-sea oceanic crust constitutes the largest region of the earth’s surface. Accumulating evidence suggests that unique microbial communities are supported by iron cycling processes, particularly in the young (<10 million-year old), cool (<25°C) subsurface oceanic crust. To test this hypothesis, we investigated the microbial abundance, diversity, and metabolic potentials in the sediment-buried crust from “North Pond” on western flank of the Mid-Atlantic Ridge. Three lithologic units along basement Hole U1383C were found, which typically hosted 65104cells cm-3of basaltic rock, with higher cell densities occurring between 115 and 145 m below seafloor. Similar bacterial community structures, which are dominated by Gammaproteobacterial and Sphingobacterial species closely related to iron oxidizers, were detected regardless of variations in sampling depth. The metabolic potentials of the crust microbiota were assayed by metagenomic analysis of two basalt enrichments which showed similar bacterial structure with the original sample. Genes coding for energy metabolism involved in hydrocarbon degradation, dissimilatory nitrate reduction to ammonium, denitrification and hydrogen oxidation were identified. Compared with other marine environments, the metagenomes from the basalt-hosted environments were enriched in pathways for Fe3+uptake, siderophore synthesis and uptake, and Fe transport, suggesting that iron metabolism is an important energy production and conservation mechanism in this system. Overall, we provide evidence that the North Pond crustal biosphere is dominated by unique bacterial groups with the potential for iron-related biogeochemical cycles.
|
| [40] |
Nitrogen stimulates the growth of subsurface basalt-associated microorganisms at the western flank of the Mid-Atlantic Ridge [J].
Oceanic crust constitutes the largest aquifer system on Earth, and microbial activity in this environment has been inferred from various geochemical analyses. However, empirical documentation of microbial activity from subsurface basalts is still lacking, particularly in the cool (<25掳C) regions of the crust, where are assumed to harbor active iron-oxidizing microbial communities. To test this hypothesis, we report the enrichment and isolation of crust-associated microorganisms from North Pond, a site of relatively young and cold basaltic basement on the western flank of the Mid-Atlantic Ridge that was sampled during Expedition 336 of the Integrated Ocean Drilling Program. Enrichment experiments with different carbon (bicarbonate, acetate, methane) and nitrogen (nitrate and ammonium) sources revealed significant cell growth (one magnitude higher cell abundance), higher intracellular DNA content, and increased Fe3+/危Fe ratios only when nitrogen substrates were added. Furthermore, aMarinobacterstrain with neutrophilic iron-oxidizing capabilities was isolated from the basalt. This work reveals that basalt-associated microorganisms at North Pond had the potential for activity and that microbial growth could be stimulated byin vitronitrogen addition. Furthermore, iron oxidation is supported as an important process for microbial communities in subsurface basalts from young and cool ridge flank basement.
|
| [41] |
Prokaryotic diversity, distribution, and insights into their role in biogeochemical cycling in marine basalts [J].
Abstract We used molecular techniques to analyze basalts of varying ages that were collected from the East Pacific Rise, 9 degrees N, from the rift axis of the Juan de Fuca Ridge and from neighboring seamounts. Cluster analysis of 16S rDNA terminal restriction fragment polymorphism data revealed that basalt endoliths are distinct from seawater and that communities clustered, to some degree, based on the age of the host rock. This age-based clustering suggests that alteration processes may affect community structure. Cloning and sequencing of bacterial and archaeal 16S rRNA genes revealed 12 different phyla and subphyla associated with basalts. These include the Gemmatimonadetes, Nitrospirae, the candidate phylum SBR1093 in the bacteria, and in the Archaea Marine Benthic Group B, none of which have been previously reported in basalts. We delineated novel ocean crust clades in the gamma-Proteobacteria, Planctomycetes and Actinobacteria that are composed entirely of basalt-associated microflora, and may represent basalt ecotypes. Finally, microarray analysis of functional genes in basalt revealed that genes coding for previously unreported processes such as carbon fixation, methane oxidation, methanogenesis and nitrogen fixation are present, suggesting that basalts harbor previously unrecognized metabolic diversity. These novel processes could exert a profound influence on ocean chemistry.
|
| [42] |
Global distribution of microbial abundance and biomass in subseafloor sediment [J].
The global geographic distribution of subseafloor sedimentary microbes and the cause(s) of that distribution are largely unexplored. Here, we show that total microbial cell abundance in subseafloor sediment varies between sites by ca. five orders of magnitude. This variation is strongly correlated with mean sedimentation rate and distance from land. Based on these correlations, we estimate global subseafloor sedimentary microbial abundance to be 2.9-10虏鈦 cells [corresponding to 4.1 petagram (Pg) C and ~0.6% of Earth's total living biomass]. This estimate of subseafloor sedimentary microbial abundance is roughly equal to previous estimates of total microbial abundance in seawater and total microbial abundance in soil. It is much lower than previous estimates of subseafloor sedimentary microbial abundance. In consequence, we estimate Earth's total number of microbes and total living biomass to be, respectively, 50-78% and 10-45% lower than previous estimates.
|
| [43] |
Meta-analysis of quantification methods shows that archaea and bacteria have similar abundances in the subseafloor [J].
There is no universally accepted method to quantify bacteria and archaea in seawater and marine sediments, and different methods have produced conflicting results with the same samples. To identify best practices, we compiled data from 65 studies, plus our own measurements, in which bacteria and archaea were quantified with fluorescent in situ hybridization (FISH), catalyzed reporter deposition FISH (CARD-FISH), polyribonucleotide FISH, or quantitative PCR (qPCR). To estimate efficiency, we defined "yield" to be the sum of bacteria and archaea counted by these techniques divided by the total number of cells. In seawater, the yield was high (median, 71%) and was similar for FISH, CARD-FISH, and polyribonucleotide FISH. In sediments, only measurements by CARD-FISH in which archaeal cells were permeabilized with proteinase K showed high yields (median, 84%). Therefore, the majority of cells in both environments appear to be alive, since they contain intact ribosomes. In sediments, the sum of bacterial and archaeal 16S rRNA gene qPCR counts was not closely related to cell counts, even after accounting for variations in copy numbers per genome. However, qPCR measurements were precise relative to other qPCR measurements made on the same samples. qPCR is therefore a reliable relative quantification method. Inconsistent results for the relative abundance of bacteria versus archaea in deep subsurface sediments were resolved by the removal of CARD-FISH measurements in which lysozyme was used to permeabilize archaeal cells and qPCR measurements which used ARCH516 as an archaeal primer or TaqMan probe. Data from best-practice methods showed that archaea and bacteria decreased as the depth in seawater and marine sediments increased, although archaea decreased more slowly.
|
| [44] |
Presence of oxygen and aerobic communities from sea floor to basement in deep-sea sediments [J].
The depth of oxygen penetration into marine sediments differs considerably from one region to another1, 2. In areas with high rates of microbial respiration, O2 penetrates only millimetres to centimetres into the sediments3, but active anaerobic microbial communities are present in sediments hundreds of metres or more below the sea floor4, 5, 6, 7. In areas with low sedimentary respiration, O2 penetrates much deeper8, 9, 10, 11, 12 but the depth to which microbial communities persist was previously unknown9, 10, 13. The sediments underlying the South Pacific Gyre exhibit extremely low areal rates of respiration9. Here we show that, in this region, microbial cells and aerobic respiration persist through the entire sediment sequence to depths of at least 75 metres below sea floor. Based on the Redfield stoichiometry of dissolved O2 and nitrate, we suggest that net aerobic respiration in these sediments is coupled to oxidation of marine organic matter. We identify a relationship of O2 penetration depth to sedimentation rate and sediment thickness. Extrapolating this relationship, we suggest that oxygen and aerobic communities may occur throughout the entire sediment sequence in 15-44% of the Pacific and 9-37% of the global sea floor. Subduction of the sediment and basalt from these regions is a source of oxidized material to the mantle.
|
| [45] |
Exploring deep microbial life in coal-bearing sediment down to ~2.5 km below the ocean floor [J].
Microbial life inhabits deeply buried marine sediments, but the extent of this vast ecosystem remains poorly constrained. Here we provide evidence for the existence of microbial communities in ~40掳 to 60掳C sediment associated with lignite coal beds at ~1.5 to 2.5 km below the seafloor in the Pacific Ocean off Japan. Microbial methanogenesis was indicated by the isotopic compositions of methane and carbon dioxide, biomarkers, cultivation data, and gas compositions. Concentrations of indigenous microbial cells below 1.5 km ranged from <10 to ~10(4) cells cm(-3). Peak concentrations occurred in lignite layers, where communities differed markedly from shallower subseafloor communities and instead resembled organotrophic communities in forest soils. This suggests that terrigenous sediments retain indigenous community members tens of millions of years after burial in the seabed.
|
| [46] |
Exploration of cultivable fungal communities in deep coal-bearing sediments from~1.3 to 2.5 km below the ocean floor [J].
Abstract Although subseafloor sediments are known to harbour a vast number of microbial cells, the distribution, diversity, and origins of fungal populations remain largely unexplored. In this study, we cultivated fungi from 34 of 47 deep coal-associated sediment samples collected at depths ranging from 1,289 to 2,457 meters below the seafloor (mbsf) off the Shimokita Peninsula, Japan (1,118 m water depth). We obtained a total of 69 fungal isolates under strict contamination controls, representing 61 Ascomycota (14 genera, 23 species) and 8 Basidiomycota (4 genera, 4 species). Penicillium and Aspergillus relatives were the most dominant genera within the Ascomycetes, followed by the members of genera Cladosporium, Hamigera, Chaetomium, Eutypella, Acremonium, Aureobasidium, Candida, Eurotium, Exophiala, Nigrospora, Bionectria and Pseudocercosporella. Four Basidiomycota species were identified as genera Schizophyllum, Irpex, Bjerkandera and Termitomyces. Among these isolates, Cladosporium sphaerospermum and Aspergillus sydowii relatives were isolated from a thin lignite coal-sandstone formation at 2,457 mbsf. Our results indicate that these cultivable fungal populations are indigenous, originating from past terrigenous environments, which have persisted, possibly as spores, through 20 million years of depositional history. This article is protected by copyright. All rights reserved. 漏 2016 Society for Applied Microbiology and John Wiley & Sons Ltd.
|
| [47] |
Endospore abundance, microbial growth and necromass turnover in deep sub-seafloor sediment [J].
Two decades of scientific ocean drilling have demonstrated widespread microbial life in deep sub-seafloor sediment, and surprisingly high microbial-cell numbers. Despite the ubiquity of life in the deep biosphere, the large community sizes and the low energy fluxes in this vast buried ecosystem are not yet understood. It is not known whether organisms of the deep biosphere are specifically adapted to extremely low energy fluxes or whether most of the observed cells are in a dormant, spore-like state. Here we apply a new approach--the D:L-amino-acid model--to quantify the distributions and turnover times of living microbial biomass, endospores and microbial necromass, as well as to determine their role in the sub-seafloor carbon budget. The approach combines sensitive analyses of unique bacterial markers (muramic acid and D-amino acids) and the bacterial endospore marker, dipicolinic acid, with racemization dynamics of stereo-isomeric amino acids. Endospores are as abundant as vegetative cells and microbial activity is extremely low, leading to microbial biomass turnover times of hundreds to thousands of years. We infer from model calculations that biomass production is sustained by organic carbon deposited from the surface photosynthetic world millions of years ago and that microbial necromass is recycled over timescales of hundreds of thousands of years.
|
| [48] |
Microbial turnover times in the deep seabed studied by amino acid racemization modelling [J].
Abstract The study of active microbial populations in deep, energy-limited marine sediments has extended our knowledge of the limits of life on Earth. Typically, microbial activity in the deep biosphere is calculated by transport-reaction modelling of pore water solutes or from experimental measurements involving radiotracers. Here we modelled microbial activity from the degree of D:L-aspartic acid racemization in microbial necromass (remains of dead microbial biomass) in sediments up to ten million years old. This recently developed approach (D:L-amino acid modelling) does not require incubation experiments and is highly sensitive in stable, low-activity environments. We applied for the first time newly established constraints on several important input parameters of the D:L-amino acid model, such as a higher aspartic acid racemization rate constant and a lower cell-specific carbon content of sub-seafloor microorganisms. Our model results show that the pool of necromass amino acids is turned over by microbial activity every few thousand years, while the turnover times of vegetative cells are in the order of years to decades. Notably, microbial turnover times in million-year-old sediment from the Peru Margin are up to 100-fold shorter than previous estimates, highlighting the influence of microbial activities on element cycling over geologic time scales.
|
| [49] |
Carbon and nitrogen assimilation in deep subseafloor microbial cells [J].
Remarkable numbers of microbial cells have been observed in global shallow to deep subseafloor sediments. Accumulating evidence indicates that deep and ancient sediments harbor living microbial life, where the flux of nutrients and energy are extremely low. However, their physiology and energy requirements remain largely unknown. We used stable isotope tracer incubation and nanometer-scale secondary ion MS to investigate the dynamics of carbon and nitrogen assimilation activities in individual microbial cells from 219-m-deep lower Pleistocene (460,000 y old) sediments from the northwestern Pacific off the Shimokita Peninsula of Japan. Sediment samples were incubated in vitro with (13)C- and/or (15)N-labeled glucose, pyruvate, acetate, bicarbonate, methane, ammonium, and amino acids. Significant incorporation of (13)C and/or (15)N and growth occurred in response to glucose, pyruvate, and amino acids (76% of total cells), whereas acetate and bicarbonate were incorporated without fostering growth. Among those substrates, a maximum substrate assimilation rate was observed at 67 脳 10(-18) mol/cell per d with bicarbonate. Neither carbon assimilation nor growth was evident in response to methane. The atomic ratios between nitrogen incorporated from ammonium and the total cellular nitrogen consistently exceeded the ratios of carbon, suggesting that subseafloor microbes preferentially require nitrogen assimilation for the recovery in vitro. Our results showed that the most deeply buried subseafloor sedimentary microbes maintain potentials for metabolic activities and that growth is generally limited by energy but not by the availability of C and N compounds.
|
| [50] |
Methyl-compound use and slow growth characterize microbial life in 2-km-deep subseafloor coal and shale beds [J].
Abstract The past decade of scientific ocean drilling has revealed seemingly ubiquitous, slow-growing microbial life within a range of deep biosphere habitats. Integrated Ocean Drilling Program Expedition 337 expanded these studies by successfully coring Miocene-aged coal beds 2 km below the seafloor hypothesized to be "hot spots" for microbial life. To characterize the activity of coal-associated microorganisms from this site, a series of stable isotope probing (SIP) experiments were conducted using intact pieces of coal and overlying shale incubated at in situ temperatures (45 C). The 30-month SIP incubations were amended with deuterated water as a passive tracer for growth and different combinations of 13 C- or 15 N-labeled methanol, methylamine, and ammonium added at low (micromolar) concentrations to investigate methylotrophy in the deep subseafloor biosphere. Although the cell densities were low (50-2,000 cells per cubic centimeter), bulk geochemical measurements and single-cell-targeted nanometer-scale secondary ion mass spectrometry demonstrated active metabolism of methylated substrates by the thermally adapted microbial assemblage, with differing substrate utilization profiles between coal and shale incubations. The conversion of labeled methylamine and methanol was predominantly through heterotrophic processes, with only minor stimulation of methanogenesis. These findings were consistent with in situ and incubation 16S rRNA gene surveys. Microbial growth estimates in the incubations ranged from several months to over 100 y, representing some of the slowest direct measurements of environmental microbial biosynthesis rates. Collectively, these data highlight a small, but viable, deep coal bed biosphere characterized by extremely slow-growing heterotrophs that can utilize a diverse range of carbon and nitrogen substrates.
|
| [51] |
Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments [J].
With the support by the National Natural Science Foundation of China,and China Ocean Mineral Resources R&D Association,the research team led by Prof.Wang Fengping(王风平)from Shanghai JiaoTong University,in cooperation with Prof.Li Meng from ShenZhen University,and Prof.Stefan M.Sievert from Woods Hole Oceanographic Institution,revealed the metabolic function of largely unknown sedi-
|
| [52] |
Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics [J].
Methanogenic and methanotrophic archaea play important roles in the global flux of methane. Culture-independent approaches are providing deeper insight into the diversity and evolution of methane-metabolizing microorganisms, but, until now, no compelling evidence has existed for methane metabolism in archaea outside the phylum Euryarchaeota. We performed metagenomic sequencing of a deep aquifer, recovering two near-complete genomes belonging to the archaeal phylum Bathyarchaeota (formerly known as the Miscellaneous Crenarchaeotal Group). These genomes contain divergent homologs of the genes necessary for methane metabolism, including those that encode the methyl-oenzyme M reductase (MCR) complex. Additional non-euryarchaeotal MCR-encoding genes identified in a range of environments suggest that unrecognized archaeal lineages may also contribute to global methane cycling. These findings indicate that methane metabolism arose before the last common ancestor of the Euryarchaeota and Bathyarchaeota.
|
| [53] |
Proceedings of the Ocean Drilling Program, Initial Reports, Vol. 139[R].Ocean Drilling Program, |
| [54] |
Integrated Ocean Drilling Program Expedition 327 Preliminary Report Juan de Fuca Ridge-Flank Hydrogeology:The Hydrogeologic Architecture of Basaltic Oceanic Crust: Compartmentalization, Anisotropy, Microbiology, and Crustal-Scale Properties on the Eastern Flank of Juan de Fuca Ridge, eastern Pacific Ocean [R]. |
| [55] |
Scientific and Technical Design and Deployment of Longterm, Subseafloor Observatories for Hydrogeologic and Related Experiments, IODP Expedition 301, Eastern Flank of Juan de Fuca Ridge[R]. |
| [56] |
|
| [57] |
Growth and population dynamics of anaerobic methane-oxidizing archaea and sulfate-reducing bacteria in a continuous-flow bioreactor [J].
The consumption of methane in anoxic marine sediments is a biogeochemical phenomenon mediated by two archaeal groups (ANME-1 and ANME-2) that exist syntrophically with sulfate-reducing bacteria. These anaerobic methanotrophs have yet to be recovered in pure culture, and key aspects of their ecology and physiology remain poorly understood. To characterize the growth and physiology of these anaerobic methanotrophs and the syntrophic sulfate-reducing bacteria, we incubated marine sediments using an anoxic, continuous-flow bioreactor during two experiments at different advective porewater flow rates. We examined the growth kinetics of anaerobic methanotrophs and Desulfosarcina-like sulfate-reducing bacteria using quantitative PCR as a proxy for cell counts, and measured methane oxidation rates using membrane-inlet mass spectrometry. Our data show that the specific growth rates of ANME-1 and ANME-2 archaea differed in response to porewater flow rates. ANME-2 methanotrophs had the highest rates in lower-flow regimes (mu(ANME-2) = 0.167 . week(-1)), whereas ANME-1 methanotrophs had the highest rates in higher-flow regimes (mu(ANME-1) = 0.218 . week(-1)). In both incubations, Desulfosarcina-like sulfate-reducing bacterial growth rates were approximately 0.3 . week(-1), and their growth dynamics suggested that sulfate-reducing bacterial growth might be facilitated by, but not dependent upon, an established anaerobic methanotrophic population. ANME-1 growth rates corroborate field observations that ANME-1 archaea flourish in higher-flow regimes. Our growth and methane oxidation rates jointly demonstrate that anaerobic methanotrophs are capable of attaining substantial growth over a range of environmental conditions used in these experiments, including relatively low methane partial pressures.
|
| [58] |
High-pressure systems for gas-phase free continuous incubation of enriched marine microbial communities performing anaerobic oxidation of methane [J].
Novel high-pressure biotechnical systems that were developed and applied for the study of anaerobic oxidation of methane (AOM) are described. The systems, referred to as high-pressure continuous incubation system (HP-CI system) and high-pressure manifold-incubation system (HP-MI system), allow for batch, fed-batch, and continuous gas-phase free incubation at high concentrations of dissolved methane and were designed to meet specific demands for studying environmental regulation and kinetics as well as for enriching microbial biomass in long-term incubation. Anoxic medium is saturated with methane in the first technical stage, and the saturated medium is supplied for biomass incubation in the second stage. Methane can be provided in continuous operation up to 20 MPa and the incubation systems can be operated during constant supply of gas-enriched medium at a hydrostatic pressure up to 45 MPa. To validate the suitability of the high-pressure systems, we present data from continuous and fed-batch incubation of highly active samples prepared from microbial mats from the Black Sea collected at a water depth of 213 m. In continuous operation in the HP-CI system initial methane-dependent sulfide production was enhanced 10- to 15-fold after increasing the methane partial pressure from near ambient pressure of 0.2 to 10.0 MPa at a hydrostatic pressure of 16.0 MPa in the incubation stage. With a hydraulic retention time of 14 h a stable effluent sulfide concentration was reached within less than 3 days and a continuing increase of the volumetric AOM rate from 1.2 to 1.7 mmol L(-1) day(-1) was observed over 14 days. In fed-batch incubation the AOM rate increased from 1.5 to 2.7 and 3.6 mmol L(-1) day(-1) when the concentration of aqueous methane was stepwise increased from 5 to 15 mmol L(-1) and 45 mmol L(-1). A methane partial pressure of 6 MPa and a hydrostatic pressure of 12 MPa in manifold fed-batch incubation in the HP-MI system yielded a sixfold increase in the volumetric AOM rate. Over subsequent incubation periods AOM rates increased from 0.6 to 1.2 mmol L(-1) day(-1) within 26 days of incubation. No inhibition of biomass activity was observed in all continuous and fed-batch incubation experiments. The organisms were able to tolerate high sulfide concentrations and extended starvation periods.
|
| [59] |
Microbial diversity and community structure of a highly active anaerobic methane-oxidizing sulfate-reducing enrichment [J].
Anaerobic oxidation of methane (AOM) is an important methane sink in the ocean but the microbes responsible for AOM are as yet resilient to cultivation. Here we describe the microbial analysis of an enrichment obtained in a novel submerged-membrane bioreactor system and capable of high-rate AOM (286 mumol g(dry weight)(-1) day(-1)) coupled to sulfate reduction. By constructing a clone library with subsequent sequencing and fluorescent in situ hybridization, we showed that the responsible methanotrophs belong to the ANME-2a subgroup of anaerobic methanotrophic archaea, and that sulfate reduction is most likely performed by sulfate-reducing bacteria commonly found in association with other ANME-related archaea in marine sediments. Another relevant portion of the bacterial sequences can be clustered within the order of Flavobacteriales but their role remains to be elucidated. Fluorescent in situ hybridization analyses showed that the ANME-2a cells occur as single cells without close contact to the bacterial syntrophic partner. Incubation with (13)C-labelled methane showed substantial incorporation of (13)C label in the bacterial C(16) fatty acids (bacterial; 20%, 44% and 49%) and in archaeal lipids, archaeol and hydroxyl-archaeol (21% and 20% respectively). The obtained data confirm that both archaea and bacteria are responsible for the anaerobic methane oxidation in a bioreactor enrichment inoculated with Eckernf枚rde bay sediment.
|
| [60] |
Bioreactor technology in marine microbiology: From design to future application [J].
Marine micro-organisms have been playing highly diverse roles over evolutionary time: they have defined the chemistry of the oceans and atmosphere. During the last decades, the bioreactors with novel designs have become an important tool to study marine microbiology and ecology in terms of: marine microorganism cultivation and deep-sea bioprocess characterization; unique bio-chemical product formation and intensification; marine waste treatment and clean energy generation. In this review we briefly summarize the current status of the bioreactor technology applied in marine microbiology and the critical parameters to take into account during the reactor design. Furthermore, when we look at the growing population, as well as, the pollution in the coastal areas of the world, it is urgent to find sustainable practices that beneficially stimulate both the economy and the natural environment. Here we outlook a few possibilities where innovative bioreactor technology can be applied to enhance energy generation and food production without harming the local marine ecosystem.
|
| [61] |
Enrichment of a microbial community performing anaerobic oxidation of methane in a continuous high-pressure bioreactor [J].
Abstract BACKGROUND: Anaerobic oxidation of methane coupled to sulphate reduction (SR-AOM) prevents more than 90% of the oceanic methane emission to the atmosphere. In a previous study, we demonstrated that the high methane pressure (1, 4.5, and 8 MPa) stimulated in vitro SR-AOM activity. However, the information on the effect of high-pressure on the microbial community structure and architecture was still lacking. RESULTS: In this study we analysed the long-term enrichment (286 days) of this microbial community, which was mediating SR-AOM in a continuous high-pressure bioreactor. 99.7% of the total biovolume represented cells in the form of small aggregates (diameter less then 15 渭m). An increase of the total biovolume was observed (2.5 times). After 286 days, the ANME-2 (anaerobic methanotrophic archaea subgroup 2) and SRB (sulphate reducing bacteria) increased with a factor 12.5 and 8.4, respectively. CONCLUSION: This paper reports a net biomass growth of communities involved in SR-AOM, incubated at high-pressure.
|
| [62] |
Single-cell genomic sequencing using multiple displacement amplification [J].
Single microbial cells can now be sequenced using DNA amplified by the Multiple Displacement Amplification (MDA) reaction. The few femtograms of DNA in a bacterium are amplified into micrograms of high molecular weight DNA suitable for DNA library construction and Sanger sequencing. The MDA-generated DNA also performs well when used directly as template for pyrosequencing by the 454 Life Sciences method. While MDA from single cells loses some of the genomic sequence, this approach will greatly accelerate the pace of sequencing from uncultured microbes. The genetically linked sequences from single cells are also a powerful tool to be used in guiding genomic assembly of shotgun sequences of multiple organisms from environmental DNA extracts (metagenomic sequences).
|
| [63] |
Manganese-and iron-dependent marine methane oxidation [J].
Anaerobic methanotrophs help regulate Earth's climate and may have been an important part of the microbial ecosystem on the early Earth. The anaerobic oxidation of methane (AOM) is often thought of as a sulfate-dependent process, despite the fact that other electron acceptors are more energetically favorable. Here, we show that microorganisms from marine methane-seep sediment in the Eel River Basin in California are capable of using manganese (birnessite) and iron (ferrihydrite) to oxidize methane, revealing that marine AOM is coupled, either directly or indirectly, to a larger variety of oxidants than previously thought. Large amounts of manganese and iron are provided to oceans from rivers, indicating that manganese- and iron-dependent AOM have the potential to be globally important.
|
| [64] |
Linking microbial phylogeny to metabolic activity at the single-cell level by using enhanced element labeling-catalyzed reporter deposition fluorescence in situ hybridization (EL-FISH) and NanoSIMS [J]. |
| [65] |
Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group [J].
The domain Archaea has historically been divided into two phyla, the Crenarchaeota and Euryarchaeota. Although regarded as members of the Crenarchaeota based on small subunit rRNA phylogeny, environmental genomics and efforts for cultivation have recently revealed two novel phyla/divisions in the Archaea; the 'Thaumarchaeota' and 'Korarchaeota'. Here, we show the genome sequence of Candidatus 'Caldiarchaeum subterraneum' that represents an uncultivated crenarchaeotic group. A composite genome was reconstructed from a metagenomic library previously prepared from a microbial mat at a geothermal water stream of a sub-surface gold mine. The genome was found to be clearly distinct from those of the known phyla/divisions, Crenarchaeota (hyperthermophiles), Euryarchaeota, Thaumarchaeota and Korarchaeota. The unique traits suggest that this crenarchaeotic group can be considered as a novel archaeal phylum/division. Moreover, C. subterraneum harbors an ubiquitin-like protein modifier system consisting of Ub, E1, E2 and small Zn RING finger family protein with structural motifs specific to eukaryotic system proteins, a system clearly distinct from the prokaryote-type system recently identified in Haloferax and Mycobacterium. The presence of such a eukaryote-type system is unprecedented in prokaryotes, and indicates that a prototype of the eukaryotic protein modifier system is present in the Archaea.
|
| [66] |
Pathways of carbon assimilation and ammonia oxidation suggested by environmental genomic analyses of marine Crenarchaeota [J].
Marine Crenarchaeota represent an abundant component of oceanic microbiota with potential to significantly influence biogeochemical cycling in marine ecosystems. Prior studies using specific archaeal lipid biomarkers and isotopic analyses indicated that planktonic Crenarchaeota have the capacity for autotrophic growth, and more recent cultivation studies support an ammonia-based chemolithoautotrophic energy metabolism. We report here analysis of fosmid sequences derived from the uncultivated marine crenarchaeote, Cenarchaeum symbiosum, focused on the reconstruction of carbon and energy metabolism. Genes predicted to encode multiple components of a modified 3-hydroxypropionate cycle of autotrophic carbon assimilation were identified, consistent with utilization of carbon dioxide as a carbon source. Additionally, genes predicted to encode a near complete oxidative tricarboxylic acid cycle were also identified, consistent with the consumption of organic carbon and in the production of intermediates for amino acid and cofactor biosynthesis. Therefore, C. symbiosum has the potential to function either as a strict autotroph, or as a mixotroph utilizing both carbon dioxide and organic material as carbon sources. From the standpoint of energy metabolism, genes predicted to encode ammonia monooxygenase subunits, ammonia permease, urease, and urea transporters were identified, consistent with the use of reduced nitrogen compounds as energy sources fueling autotrophic metabolism. Homologues of these genes, recovered from ocean waters worldwide, demonstrate the conservation and ubiquity of crenarchaeal pathways for carbon assimilation and ammonia oxidation. These findings further substantiate the likely global metabolic importance of Crenarchaeota with respect to key steps in the biogeochemical transformation of carbon and nitrogen in marine ecosystems.
|
| [67] |
Illuminating Earth’s Past, Present and Future. The Science Plan for the International Ocean Discovery Program 2013-2023[R] . |
| [68] |
Spatial variations in microbial community composition in surface seawater from the ultra-oligotrophic center to rim of the South Pacific Gyre [J].
Surface seawater in the South Pacific Gyre (SPG) is one of the cleanest oceanic environments on earth, and the photosynthetic primary production is extremely low. Despite the ecological significance of the largest aquatic desert on our planet, microbial community composition in the ultra-oligotrophic seawater remain largely unknown. In this study, we collected surface seawater along a southern transect of the SPG during the Integrated Ocean Drilling Program (IODP) Expedition 329. Samples from four distinct sites (Sites U1368, U1369, U1370 and U1371) were examined, representing 655400 kilometers of transect line from the gyre heart to the edge area. Real-time PCR analysis showed 16S rRNA gene abundance in the gyre seawater, ranging from 5.96×105 to 2.55×106 copies ml611 for Bacteria and 1.17×103 to 1.90×104 copies ml611 for Archaea. The results obtained by statistic analyses of 16S rRNA gene clone libraries revealed the community composition in the southern SPG area: diversity richness estimators in the gyre center (Sites U1368 & U1369) are generally lower than those at sites in the gyre edge (Sites U1370 & U1371) and their community structures are clearly distinguishable. Phylogenetic analysis showed the predominance of Proteobacteria (especially Alphaproteobacteria) and Cyanobacteria in bacterial 16S rRNA gene clone libraries, whereas phylotypes of Betaproteobacteria were only detected in the central gyre. Archaeal 16S rRNA genes in the clone libraries were predominated by the sequences of Marine Group II within the Euryarchaeota, and the Crenarchaeota sequences were rarely detected, which is consistent with the real-time PCR data (only 9.9 to 22.1 copies ml611). We also performed cultivation of heterotrophic microbes onboard, resulting in 18.9% of phylogenetically distinct bacterial isolates at least at the species level. Our results suggest that the distribution and diversity of microbial communities in the SPG surface seawater are closely related to the ultra-oligotrophic oceanographic features in the Pacific Ocean.
|
| [69] |
Succession of bacterial community structure and potential significance along a sediment core from site U1433 of IODP expedition 349, South China Sea [J].
To evaluate the potential impact of geological processes and depositional history on shaping the subsurface biosphere, the bacterial community structures in a sediment core of the South China Sea was investigated by molecular approaches that target 16S rRNA gene fragments. Samples were obtained from different lithologic intervals at site U1433 during the International Ocean Discovery Program (IODP) Expedition 349. Bacterial abundance decreased rapidly with depth, with nearly three orders of magnitude decline within the first 10002m below seafloor (mbsf). Community diversity displayed a similar decreasing pattern, yet, a slight increase in diversity emerged in the early to middle Miocene. Such excursion might reflect enhanced cell activity in response to increasing temperature due to a steep geothermal gradient. Non-metric multidimensional scaling ordination revealed that the bacterial communities along the sediment core represent four clusters based on depth and geological time. There were distinct bacterial community shifts among clusters at 4.50–98.9302mbsf (Pleistocene), 108.15–273.2002mbsf (Pleistocene), 296.09–709.1302mbsf (Pliocene and late Miocene), and 732.10–789.9102mbsf (early to middle Miocene). Classification analysis revealed a striking pattern: the relative abundance of microorganisms affiliated with Gammaproteobacteria , Actinobacteria , and Cyanobacteria overall consistently decreased with depth, whereas those affiliated with Chloroflexi , candidate division OP9, candidate phylum BHI80-139, and Nitrospirae increased; these findings correspond to different clusters. Total organic carbon content and ratio of total organic carbon: total nitrogen, along with pore water phosphate concentration and salinity, were the statistically most significant variables that explained the bacterial community cluster pattern, which indicates potential linkages of bacterial communities to changes in quality and quantity of buried organic matter over geological time scales. Geographic isolation across depth was more important than environmental condition and geological age for the development of unique community structure in marine deep biosphere, although environmental variables partially shaped bacterial community composition.
|
| [70] |
Predominance of Viable Spore-Forming Piezophilic Bacteria in High-Pressure Enrichment Cultures from ~1.5 to 2.4 km-Deep Coal-Bearing Sediments below the Ocean Floor [J].
Phylogenetically diverse microorganisms have been observed in marine subsurface sediments down to ~2.5 km below the seafloor (kmbsf). However, very little is known about the pressure-adapted and/or pressure-loving microorganisms, the so called piezophiles, in the deep subseafloor biosphere, despite that pressure directly affects microbial physiology, metabolism and biogeochemical processes of carbon and other elements in situ. In this study, we studied taxonomic compositions of microbial communities in high-pressure cultivated sediment, obtained during the Integrated Ocean Drilling Program (IODP) Expedition 337 off the Shimokita Peninsula, Japan. Analysis of 16S rRNA gene-tagged sequences showed that members of spore-forming bacteria within Firmicutes and Actinobacteira were predominantly detected in all enrichment cultures from ~1.5 to 2.4 km-deep sediment samples, with the sequence frequency followed by members of Proteobacteria, Acidobacteria, and Bacteroidetes. To further study the physiology of the deep subseafloor sedimentary piezophilic bacteria, we isolated and characterized two bacterial strains, 19R1-5 and 29R7-12, from 1.9 and 2.4 km-deep sediment samples, respectively. The isolates were both low G+C content, gram-positive, endospore-forming and facultative anaerobic piezophilic bacteria, closely related to Virgibacillus pantothenticus and Bacillus subtilis within the phylum Firmicutes, respectively. The optimal pressure and temperature conditions for growth were 20 MPa and 42oC for strain 19R1-5, and 10 MPa and 43oC for strain 29R7-12. Bacterial (endo)spores were observed in both the enrichment and pure cultures examined, suggesting that these piezophilic members were derived from microbial communities buried in the ~20 million-year-old coal-bearing sediments after the long-term survival as spores and that the deep biosphere may host more abundant gram-positive spore-forming bacteria and their spores than hitherto recognized.
|
| [71] |
Microbial sulfur cycle in two hydrothermal chimneys on the Southwest Indian Ridge [J].
Sulfur is an important element in sustaining microbial communities present in hydrothermal vents. Sulfur oxidation has been extensively studied due to its importance in chemosynthetic pathways in hydrothermal fields; however, less is known about sulfate reduction. Here, the metagenomes of hydrothermal chimneys located on the ultraslow-spreading Southwest Indian Ridge (SWIR) were pyrosequenced to elucidate the associated microbial sulfur cycle. A taxonomic summary of known genes revealed a few dominant bacteria that participated in the microbial sulfur cycle, particularly sulfate-reducing Deltaproteobacteria. The metagenomes studied contained highly abundant genes related to sulfur oxidation and reduction. Several carbon metabolic pathways, in particular the Calvin-Benson-Bassham pathway and the reductive tricarboxylic acid cycles for CO2 fixation, were identified in sulfur-oxidizing autotrophic bacteria. In contrast, highly abundant genes related to the oxidation of short-chain alkanes were grouped with sulfate-reducing bacteria, suggesting an important role for short-chain alkanes in the sulfur cycle. Furthermore, sulfur-oxidizing bacteria were associated with enrichment for genes involved in the denitrification pathway, while sulfate-reducing bacteria displayed enrichment for genes responsible for hydrogen utilization. In conclusion, this study provides insights regarding major microbial metabolic activities that are driven by the sulfur cycle in low-temperature hydrothermal chimneys present on an ultraslow midocean ridge.There have been limited studies on chimney sulfides located at ultraslow-spreading ridges. The analysis of metagenomes of hydrothermal chimneys on the ultraslow-spreading Southwest Indian Ridge suggests the presence of a microbial sulfur cycle. The sulfur cycle should be centralized within a microbial community that displays enrichment for sulfur metabolism-related genes. The present study elucidated a significant role of the microbial sulfur cycle in sustaining an entire microbial community in low-temperature hydrothermal chimneys on an ultraslow spreading midocean ridge, which has characteristics distinct from those of other types of hydrothermal fields.
|
| [72] |
Complete genome sequence and whole-genome phylogeny of Kosmotoga pacifica type strain SLHLJ1 T from an East Pacific hydrothermal sediment [J].
Kosmotoga pacificastrain SLHLJ1Tis a thermophilic chemoorganoheterotrophic bacterium isolated from a deep-sea hydrothermal sediment. It belongs to the physiologically homogeneousThermotogaceaefamily. Here, we describe the phenotypic features ofK. pacificatogether with its genome sequence and annotation. The chromosome has 2,169,170聽bp, organized in one contig. A total of 1897 candidate protein-encoding genes and 177 RNA genes were identified. The 16S rRNA gene sequence of this strain is distantly related to sequences of some relatives classified in the same genus (K. olearia7.02% andK. shengliensis7.83%), with dissimilarity percentages close to the threshold generally described for genus delineation. Nevertheless, the percentage of conserved proteins (POCP), which is much higher than 50% (around 70%), together with phenotypic features of the isolates, confirm the affiliation allKosmotogaspecies described so far to the same genus. The online version of this article (doi:10.1186/s40793-016-0214-2) contains supplementary material, which is available to authorized users.
|
| [73] |
Diversity of culturable sulfur-oxidizing bacteria in deep-sea hydrothermal vent environments of the South Atlantic [J].
[Objective] To investigate the diversity of culturable sulfur-oxidizing bacteria in hydrothermal vent environments of the South Atlantic, and analyze their characteristics of sulfur oxidation. [Methods] We enriched and isolated sulfur-oxidizing bacteria from hydrothermal vent samples collected from the South Atlantic. The microbial diversity in enrichment cultures was analyzed using the Denatural Gradient Gel Electrophoresis method. Sulfur-oxidizing characteristics of the isolates was further studied by using ion chromatography. [Results] A total of 48 isolates were obtained from the deep-sea hydrothermal vent samples, which belonged to 23 genera and mainly grouped into alphaProteobacteria(58.3%), Actinobacteria(22.9%) and gama-Proteobacteria(18.8%). Among them, the genus Thalassospira,Martelella and Microbacterium were dominant. About 60% of the isolates exibited sulfur-oxidizing ability and strain L6M1-5 had a higher sulfur oxidation rate by comparison analysis. [Conclusion] The diversity of sulfur-oxidizing bacteria in hydrothermal environments of the South Atlantic was reported for the first time based on culture-dependent methods. The result will help understand the biogechemical process of sulfur compounds in the deep-sea hydrothermal environments.
|
| [74] |
Thermophilic hydrogen-producing bacteria inhabiting deep-sea hydrothermal environments represented by Caloranaerobacter [J].
Hydrogen is an important energy source for deep-sea hydrothermal vent ecosystems. However, little is known about microbes and their role in hydrogen turnover in the environment. In this study, the diversity and physiological characteristics of fermentative hydrogen-producing microbes from deep-sea hydrothermal vent fields were described for the first time. Seven enrichments were obtained from hydrothermal vent sulfides collected from the Southwest Indian Ocean, East Pacific and South Atlantic. 16S rRNA gene analysis revealed that members of the Caloranaerobacter genus were the dominant component in these enrichments. Subsequently, three thermophilic hydrogen producers, strains H363, H53214 and DY22619, were isolated. They were phylogenetically related to species of the genus Caloranaerobacter. The H2 yields of strains H363, H53214, DY22619 and MV107, which was the type species of genus Caloranaerobacter, were 0.11, 1.21, 3.13 and 2.85聽mol H2/mol glucose, respectively. Determination of the main soluble metabolites revealed that strains H363, H53214 and MV107 performed heterolactic fermentations, while strain DY22619 performed butyric acid fermentation, indicating distinct fermentation patterns among members of the genus. Finally, a diversity of forms of [FeFe]-hydrogenase with different modular structures was revealed based on draft genomic data of Caloranaerobacter strains. This highlights the complexity of hydrogen metabolism in Caloranaerobacter, reflecting adaptations to environmental conditions in hydrothermal vent systems. Collectively, results suggested that Caloranaerobacter species might be ubiquitous and play a role in biological hydrogen generation in deep-sea hydrothermal vent fields.
|
| [75] |
Thermococcus eurythermalis sp. nov., A conditional piezophilic, hyperthermophilic archaeon with a wide temperature range for growth, isolated from an oil-immersed chimney in the Guaymas Basin [J].
A conditional piezophilic, hyperthermophilic archaeon showing growth over a wide range of temperature, pH and pressure was isolated from an oil-immersed hydrothermal chimney at a depth of 2006.9 m in the Guaymas Basin. Enrichment and isolation of strain A501(T) were performed at 80 °C at 0.1 MPa. Cells of isolate A501(T) were irregular motile cocci with a polar tuft of flagella and generally 0.6-2.6 08m in diameter. Growth was detected over the range 50-100 °C (optimal growth at 85 °C) at atmospheric pressure and was observed at 102 °C at a pressure of 10 MPa. At 85 °C, growth was observed at a pressure of 0.1-70 MPa (optimum pressure 0.1 MPa-30 MPa), while at 95 °C, the pressure allowing growth ranged from 0.1 MPa to 50 MPa (optimum pressure 10 MPa). Cells of strain A501(T) grew at pH 4-9 (optimum pH 7.0) and a NaCl concentration of 1.0-5.066% (w/v) (optimum concentration 2.566% NaCl). This isolate was an anaerobic chemo-organoheterotroph and was able to utilize yeast extract, peptone, tryptone and starch as the single carbon source for growth. Elemental sulfur and cysteine stimulated growth; however, these molecules were not necessary. The DNA G+C content of the complete genome was 53.47 mol%. The results of 16S rRNA gene sequence analysis indicated that strain A501(T) belongs to the genus Thermococcus. There was no significant similarity between strain A501(T) and the phylogenetically related species of the genus Thermococcus based on complete genome sequence alignments and calculation of the average nucleotide identity and the tetranucleotide signature frequency correlation coefficient. These results indicate that strain A501(T) represents a novel species, Thermococcus eurythermalis sp. nov. The type strain is A501(T) (66=66CGMCC 7834(T)66=66JCM 30233(T)).
|
| [76] |
Complete genome sequence of Thermococcus eurythermalis A501, a conditional piezophilic hyperthermophilic archaeon with a wide temperature range, isolated from an oil-immersed deep-sea hydrothermal chimney on Guaymas Basin [J]. |
| [77] |
Current developments in marine microbiology: High-pressure biotechnology and the genetic engineering of piezophiles [J].
Abstract A key aspect of marine environments is elevated pressure; for example, 70% of the ocean is at a pressure of at least 38MPa. Many types of Bacteria and Archaea reside under these high pressures, which drive oceanic biogeochemical cycles and catalyze reactions among rocks, sediments and fluids. Most marine prokaryotes are classified as piezotolerant or as (obligate)-piezophiles with few cultivated relatives. The biochemistry and physiology of these organisms are largely unknown. Recently, high-pressure cultivation technology has been combined with omics and DNA recombination methodologies to examine the physiology of piezophilic marine microorganisms. We are now beginning to understand the adaptive mechanisms of these organisms, along with their ecological functions and evolutionary processes. This knowledge is leading to the further development of high-pressure-based biotechnology. Copyright 漏 2015 Elsevier Ltd. All rights reserved.
|
| [78] |
Life in a multi-extreme environment: Thermococcales living in deep sea hydrothermal vents [J].
Thermococcales are commonly the dominant microbesin the black chimneys of deep sea hydrothermal vents, which representone of the most extreme environments on earth and are similar to theconditions of the early earth. Thermococcales haveadapted to the dramatic environmental fluctuations of physical andchemical factors in hydrothermal vents. They usually have wide growthranges (over 40掳C for temperature, over 5 pH units for pH, andover 80-塎Pa for pressure growth ranges) but small genome sizes(<2.3-塎b) compared with most microorganisms. Studies onthe adaptation of Thermococcales to various extremeenvironments have reported that some of their unique metabolic pathwaysare related to adaptation to multiple extreme environments. Thesemetabolic pathways include compatible solute, energetic metabolism,membrane lipid component, amino acid metabolism, and antioxidationpathways. Exploring these pathways further revealed that Thermococcales may have common adaptation strategies to multiple extreme environments.Studying the common adaptation mechanisms of Thermococcales will reveal the survival strategies of microorganisms in low-energeticand high-temperature extreme environments such as deep biospheresor extraterritorial habitats. It will also provide valuable researchmodels and ideas for investigating metabolic traits of early lifeand further enhance our understanding of the origin of life. Finally,it will also provide theoretic references and biological materialsfor use in synthetic biology research and industrial applications.
|
| [79] |
Methane-metabolizing microbial communities in sediments of the Haima cold seep area, northwest slope of the South China Sea [J].
react-text: 146 Dimethylsulfide (DMS) and dimethylsulfoniopropionate (DMSP) production by Scrippsiella trochoidea and Prorocentrum minimum was investigated to characterize the effects of physiological stage and salinity on DMS and DMSP pools of these two marine phytoplankton species. Axenic laboratory cultures of the two marine algae were tested for DMSP production and its conversion into DMS. The results... /react-text react-text: 147 /react-text [Show full abstract]
|
| [80] |
Ecosystems of cold seeps in the South China Sea [C]∥ |

/
| 〈 |
|
〉 |

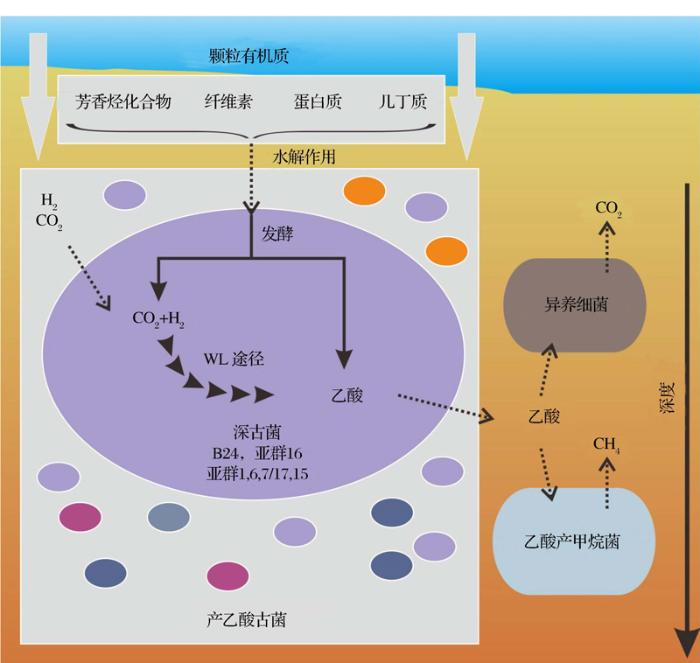
{kind=link}
{kind=link}