1 引言
天然放射性碳同位素(14 C)主要来源于高层大气层中宇宙射线产生的中子与稳定氮同位素14 N的反应,产生的14 C与大气中的氧结合形成二氧化碳(14 CO2 )进入低层大气层,通过海—气交换和垂直混合进入海洋,成为陆地和海洋生物生命活动(如光合作用和化能自养等的同化作用)的无机碳源。可见,14 C广泛存在于大气、陆地、海洋及生物体中,这使得14 C成为了解生物地球化学碳循环过程和研究各个碳库之间碳交换的有力的示踪工具。另外,人类核试验也是大气层中14 C的重要来源。尤其是20世纪50~60年代密集的核试验,使得北半球大气层的14 C浓度增加了1倍,60年代初期,对流层中CO2 的Δ14 C值超过了900‰[1 ] 。自1962年全面禁止核试验以来,大气中由核试验引入的14 CO2 被海洋和陆地生物吸收不断地衰变减少,通过环境生物样品核爆炸标记(“bomb-spike”)的Δ14 C信号的变化,可作为短时间尺度生物地球化学过程的同位素示踪剂[2 ] 。
作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学。其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] 。在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响。有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] 。但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年。例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] 。在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] 。Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平。此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄。由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响。因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年。尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] 。
另外,生物标志物单体14 C同位素也是一类常见的来源示踪指标,被广泛用于大气颗粒物、土壤、河流、湖泊、海洋等生态环境系统中有机碳的来源解析研究中[15 ~20 ] 。由于高的初级生产力以及高的陆源输入,边缘海不仅是重要的海洋碳汇,也是一个重要的陆源碳汇。有机碳储库(carbon pools)(颗粒有机碳汇和溶解有机碳汇)中不同来源有机质的比例不仅决定了其组成性质和埋藏效率,而且还在不同尺度上影响全球碳循环。通过生物标志物单体14 C同位素技术可以将自然环境系统中有机质来源从“年龄”的角度基本可划分为三大类,包括年轻的海源有机质和陆源维管植物残留物(Δ14 C=0±50‰,近现代: <100年)、陈化的土壤有机质(千年尺度)及古老的沉积岩风化有机质(Δ14 C=-1 000‰,万年尺度)[21 ,22 ] 。以上3种有机质中只有海洋生源和植被源有机碳汇能够影响短时间尺度大气CO2 的浓度,而陈化土壤源和化石源古老有机质常常会在陆地湖泊、土壤或沉积岩等中间有机碳储库中埋藏成百上千年甚至百万年,随时间的推移,通过河流或者风尘等传输过程进入海洋沉积环境中,所以这些陆源有机质的埋藏在短时间尺度上对近现代大气CO2 的埋藏贡献几乎为零。因此,通过14 C同位素特征将现代有机碳和其他年龄的有机碳区分开,有助于正确评估现代海洋“碳汇”的功能,并从“年龄”的新视角认识有机碳的迁移、转化和埋藏特征。大部分自然样品中有机碳的来源复杂,这一异质性导致总有机质的表观14 C同位素组成代表的是一个混合的信号。尤其是陈化土壤有机质的Δ14 C端元值的高度时空可变性[23 ,24 ] ,很难通过在自然界中找到对应的典型样品来确定它,导致总有机质14 C同位素信号无法区分和剥离较老的土壤源有机质和古老的化石源有机质的信号。由于有机物的14 C同位素特征不依赖于生物合成方式和化合物种类,只受合成时利用的碳源的14 C同位素水平和“中间储库”停留时间控制。生物标志物由特定生物合成,性质稳定,在分子水平上指示其特定来源[25 ] ,生物标志物14 C同位素组成被广泛应用于评价生物圈中有机质的来源及组成变化[16 ,26 ~31 ] 。因此,通过来源具有专一性(Source Specific Characteristics)不同生物标志物单体14 C信号可以准确获取相应来源有机质的14 C同位素特征端元值,以达到示踪这类有机物“源—汇”过程变化。
Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA)。CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量。通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河。此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中。近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] 。对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] 。因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] 。除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] 。本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状。
2 化学分离提纯技术
自然环境研究中常用生物标志物的含量一般很低(一般小于1 μg/g干重,甚至低于几十个ng/g干重),所以需要通过分离和富集从自然环境样品中获取足够量(>100 μg C)、高纯度的生物标志物以满足AMS的测试要求。为了消除天然环境样品中复杂的基质干扰,首先会利用湿化学法进行目标组分初步的有机溶剂萃取和分离提纯。有机溶剂萃取技术包括超声萃取[44 ,45 ] 、索氏提取[27 ,46 ] 、加速溶剂萃取[47 ,48 ] 以及微波萃取[14 ,17 ] 等技术。在提纯生物标志物过程中,还需要配合一系列的化学分离技术和固相萃取技术等手段。根据化合物的不同化学性质,如中性组分(包括烷烃、醇类和烯酮等)和酸性组分(包括脂肪酸等),可以通过碱水解(0.5 mol/L的KOH的甲醇溶液)将二者分离[49 ] 。固相萃取分离技术则是根据不同目标化合物的极性或结构差异实现进一步分离和提纯的化学法。例如,硅胶层析通过改变淋洗液极性梯度,可将不同极性的化合物分离[49 ] ;硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] 。然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体。
3 传统PCGC技术
天然样品的CSRA分析中,PCGC是最早发展起来的用于同位素分析的高纯度单体制备色谱技术。PCGC具有高的分离能力,克服了毛细管气相色谱的有限容量(<500 ng/compound,compound为单个化合物)的技术限制。整个装置由2部分组成:气相色谱和Gerstel八通阀回收装置,共6个回收窗口(图1 )。气相色谱端耦合了CIS系统程序升温的PTV进样口、大口径色谱柱(0.53 mm(内径,i, d),0.5 μm(固定相膜厚,film thickness))、FID检测器、十字分流阀。溶剂排空方式的PTV进样口和大孔径色谱柱的使用可实现大体积(≥5 μL/injection)、大容量(约2 μg/compound)进样,通过30~50次的重复进样可以收集足够量的目标化合物(60~100 μg/compound)满足现代高灵敏度AMS[50 ,51 ] 的14 C同位素测定需要。经过高效气相色谱柱分离的目标有机化合物单体通过十字分流阀,约1%进入FID检测器,其余99%通过毛细管传输线(transfer line,0.32 mm(内径,i.d.))进入Gerstel八通阀回收装置;低的保留时间漂移程度(标准偏差最大为0.03 min)保证目标化合物多次收集的准确性和可靠性[26 ] 。Eglinton等[26 ] 首次应用PCGC分离富集了海洋沉积物中常见的类脂生物标志物单体,实现了自然环境浓度水平的单体14 C测定。然而CSRA技术仍然遇到很多的挑战和限制,导致其还没有真正实现全自动化,甚至相关的质量控制标准也未建立。在自然环境研究中,CSRA技术的应用所面临的挑战和瓶颈主要是环境样品中常见的几类生物标志物含量都较低,其有机化合物单体的纯化制备过程复杂繁琐且技术要求高。随着高灵敏度加速器质谱的发展,14 C同位素测定所需的样品量越来越小,甚至能够测试最低至几个微克碳的样品[36 ,52 ] 。因此,过程空白或外源碳(Cex )对样品尤其是小样品最终14 C结果产生的影响就愈加明显[37 ,40 ,53 ] 。
图1 PCGC系统工作原理图示[26 ]
Fig.1 Diagrammatic representation of the PCGC instrument[26 ]
3.1 外源碳(Cex )或过程空白的控制
从自然环境样品中分离富集获取高纯度目标生物标志物单体进行14 C同位素测定,需要进行化学萃取过程、PCGC分离过程、CO2 气体定量/石墨化过程、AMS测定等,以上每一个步骤中都有可能将来源和同位素组成不明的外源碳引入至CSRA样品中,严格的空白评估和质量控制将是放射性碳同位素结果准确性和可靠性的重要保障,尤其是超微样品(<25 μg C)。
3.1.1 实验环境条件的控制
首先,保证一个无14 C-标记的实验环境是进行自然水平14 C浓度水平分析测试的前提。因为即使很少的14 C-标记化合物,也足以对放射性碳同位素分析造成非常大的偏差。 14 C/12 C的自然含量仅为1.2×10-12 ,即使3 × 10-18 ng的14 C也可以使25 μg C样品的Δ14 C值发生很大的改变(从+100‰到+1100‰)[54 ,55 ] ,并且14 C-标记的化合物不能通过GC-FID等技术手段进行快速的监测。不过,尽管14 C-标记化合物是14 C分析中最为严重的外源碳污染,但是严格的实验室管理措施可以避免添加14 C的污染[55 ] 。
3.1.2 PCGC分离过程空白的评估
随着CSRA技术在自然环境样品中的应用,样品量逐渐向超微样品(<25 μg C)发展,此时过程空白也就成为了CSRA样品14 C测试精确度和准确度的实际限制因素[37 ,56 ] 。自然环境中的样品,化学萃取和PCGC分离富集过程中引入的外源碳是过程空白的主要贡献,远远高于在真空线转换为CO2 和/或石墨化过程中外源碳的量;并且当样品量变小时(<100 μg C),过程空白的评价显得更为重要[40 ] 。在PCGC分离过程中,色谱柱流失是外源碳的主要来源之一。Eglinton等[26 ] 发现,当PCGC使用高容量色谱柱(>1 μm固定相膜厚)时,尤其是高沸点的有机化合物单体组分中,GC-FID能够明显从PCGC-U型捕集管中检测出聚甲基硅酮的降解产物;而对于使用较薄的固定相时(≤0.5 μm),色谱柱流失的影响则明显减少。
定量估算PCGC分离过程所引入外源碳的方法分别为直接法(directly method)和间接法(indirectly method)[38 ,57 ] 。直接法是假定在PCGC分离过程中的外源碳主要是来自柱流失(包括过去样品的残留和/或GC色谱柱固定相的脱落),且柱流失不随时间和温度的变化而改变。在无溶剂条件下运行PCGC,馏分收集器按照目标化合物的截留时间收集相同条件下的仪器本底空白。Ziolkowski等[38 ] 通过直接法评估了PCGC分离过程中空白的质量随馏分收集器的收集窗口时间增加而增加,平均产生外源碳为(0.1±0.05) μg C /min(Fm=0.125±0.034)。而间接方法则是通过加入已知14 C同位素水平的参考标准——“现代源(modern)”有机化合物标准(Fm=1)和“化石源(fossil)”有机化合物标准(Fm=0),分别评估PCGC分离过程中引入的化石源空白碳和现代空白碳的质量。二者质量加和即为引入的外源碳空白总质量,二者Fm的质量加权平均值即为外源碳空白的同位素特征值[38 ] 。Ziolkowski等[38 ] 的研究结果显示通过间接法评估得到的PCGC分离过程中引入的外源碳中化石源空白碳和现代源空白碳的贡献分别是Cpcgc-fossil =(0.4±0.2) μg C/min和Cpcgc-modern =(0.2±0.1) μg C/min。因此,应用间接方法估算的外源碳的贡献是Cpcgc =(0.6±0.3) μg C/min(Fm=0.3±0.1)。2种方法估算的PCGC分离过程中引入外源碳的贡献相差较大(0.5 μg C),这可能是在有溶剂条件下,来自色谱柱的外源碳更容易被洗脱以及样品的记忆效应和/或进样口的污染可能也是间接法呈现较高过程空白的影响因素。但Coppola等[57 ] 的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素。Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献。由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源。但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小。另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源。由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献。前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况。这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] 。通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失。对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除。另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] 。Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程。通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] 。陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13)。由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估。
另外,稳定碳同位素手段也可以用来初步鉴别CSRA是否受到明显外来碳的影响。假定外源碳和目标化合物的13 C值有明显的差别,通过比较稳定碳同位素比质谱仪测定的目标有机物单体δ13 C值和PCGC分离富集回收后的目标化合物的δ13 C值之间的差异,也可以评估是否存在有机溶剂残留或者是其他污染外源碳被引入[26 ,54 ] 。
3.2 同位素分馏效应
由于PCGC分离过程中目标化合物在高温条件下以气态的形式传输后进入捕集管,整个系统气密性的可持续性决定了目标化合物回收率的好坏,实际实验过程中PCGC的回收率一般在70%~80%[26 ,60 ] 。在色谱分离过程中通常会发生同位素分馏,随着色谱信号的流出,其碳同位素组成是不断变化的[61 ,62 ] 。因此,PCGC分离富集过程中目标化合物不完全回收往往也会导致一定的碳同位素分馏。Zencak等[54 ] 测定了香草醛标准不同截留时间馏分的δ13 C,结果显示其谱峰的前沿部分(-15.75‰)明显偏正于峰尾部分(-49.91‰),这与Eglinton等[26 ] 的结果一致。碳同位素呈现逆同位素效应(inverse isotope effect),即重的碳同位素先流出而轻的同位素后流出,这与Cl等同位素效应相反[63 ] 。因此当目标化合物不完全分离或收集,会发生较为明显的碳同位素分馏。Zencak等[54 ] 同时指出,未经处理的香草醛δ13 C比值(-31.6‰)与经PCGC完整收集的香草醛δ13 C比值(-31.3‰)一致。但实际中,尤其是处理海洋沉积物等基质复杂的样品,为了获取高纯度的目标化合物,通常会缩短组分截留窗口的时间,即靠近目标化合物拖尾或前沿的部分截留,这可能导致目标化合物不完全回收而引起一定程度的碳同位素分馏。理论上,δ14 C的同位素分馏应当是δ13 C分馏的2倍,呈现和13 C同样的逆同位素效应。但是,放射性碳数据通常是报道经过δ13 C=-25‰校正后的Fm和Δ14 C值[64 ] 。因此,即使发生碳同位素分馏,只会影响目标化合物的回收率及δ13 C数据的准确性,而不会影响最终报道的放射性碳同位素数据[64 ] 。
4 二维PCGC技术
传统的PCGC配置了大孔径色谱柱(0.53 mm i.d.),提供了大负载容量的同时,牺牲了色谱的分离能力[43 ] 。而为了弥补传统PCGC分离能力且减少或避免使用可能引入外源碳的其他湿化学萃取分离过程[38 ,54 ,57 ] ,有报道指出可以通过串联色谱及多维色谱技术提高传统PCGC对复杂基质的分离能力,同时也减少了繁琐的化学分离前处理过程中潜在外源碳的引入[42 ] 。Ball等[42 ] 将二维PCGC应用于CSRA样品的分离。与传统PCGC相比,其增加了一个中心切割微流体切换装置(Deans pneumatic microfluidic switching device)以及串联2个不同固定相的色谱柱(Rtx-5 MS, 0.50 μm df和DB-17 MS, 0.25 μm df)。尽管增加了微流体切换装置及色谱柱数量,但其保留时间漂移程度很小(标准偏差最大为0.016 min),这与传统PCGC的性能基本一致[26 ] 。而多次进样发现其FID峰面积的偏差为1.1%~1.2%[42 ] ,这可能是与PTV进样模式所引起的扰动有关。传统PCGC柱流失对过程空白的贡献是(0.6±0.3) μg C/min,但如果无溶剂空运行PCGC,则柱流失仅贡献了0.1 μg C/min[38 ] 。而在二维PCGC中,2°色谱柱仅仅从1°柱中接收有限量的样品,而并没有溶剂进入2°柱,因此柱流失并不是二维PCGC技术外源碳的重要来源。另外,在二维PCGC中使用了较薄的(0.25 μm, df)和低柱流失的固定相,可以进一步降低柱流失的贡献。Ball等[42 ] 通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定。
5 Prep-HPLC技术
Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段。Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致。可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标。Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右。但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] 。同时,衍生化试剂也会引入一定的外源碳[65 ] 。为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试。
利用半制备型氨基柱和分析型氨基柱的2步分离的Prep-HPLC技术被成功用于分离海洋沉积物中的GDGTs、植醇及甾醇等生物标志物[69 ] 。该Prep-HPLC系统耦合了流动注射分析技术、大气压电离质谱、HP-1100液相色谱(HPLC)和馏分收集装置。根据液相色谱柱分离后的目标化合物单体进入大气压电离质谱(APCI-MS)检测器分子离子信号峰的保留时间设定馏分收集器的截留时间。化学分离纯化后的极性类脂组分(见本文第2部分介绍)通过半制备型氨基柱(Econosphere, 250 mm×10 mm, 10 μm; Alltech Associates)进行初步的分离收集,然后使用分析型氨基柱(Econosphere, 250 mm×4.6 mm, 5 μm; Alltech Associates)进一步分离纯化。Smittenberg等[69 ] 通过GC,GCMSD,HPLC-MS和核磁共振(13 C-NMR和1 H-NMR)手段分别对以上Prep-HPLC分离富集所得的甾醇类和GDGT(GDGT-0和Crenarcheaol)类有机化合物单体进行了纯度检测,类脂有机化合物单体纯度高于95%,且没有其他明显的外源碳的污染。与PCGC相比较,具有相对高的回收效率(>89%)。Prep-HPLC成功地分离自然环境样品中高极性、高沸点(>C30)生物标志物,扩充生物标志物单体14 C的应用范围。此外,Prep-HPLC具有更大的柱容量(高达30 μg/injection),相对于PCGC分离富集的时间大大缩短。
Birhkolz等[48 ] 改进了已有的Prep-HPLC技术,成功分离富集出湖泊沉积物中br-GDGTs化合物单体。首先利用反相-HPLC(Agilent Eclipse Plus C18 column,4.6 mm×150 mm, 3.5 μm)将br-GDGTs和共流出组分分离;为了除去反相-HPLC柱分离过程中引入的共流出组分和进一步纯化,使用正相-HPLC柱(Alltech Prevail Cyano column, 2.1 mm×150 mm, 3 μm)消除可能来自流动相的外源碳(12 μg C/60 min)。改进后的HPLC技术贡献的外源碳为 (0.6±0.2) μg C(其F14 C为0.5±0.2),与Hou等[14 ] 结果基本一致,其外源碳的贡献约为2 μg C(Fm值为0.30)。基于br-GDGTs的BIT指标已被广泛用来估算海洋环境中土壤有机质的贡献比例[71 ~73 ] ,但有报道指出湖泊[74 ] 和海洋[75 ] 也会产生br-GDGTs。湖泊、海洋环境样品中br-GDGTs的14 C同位素特征可以为br-GDGTs土壤源指标的应用提供重要的信息。Birkholz等[48 ] 发现Lake Lusvatnet沉积物中的br-GDGTs呈现较老的14 C年龄。结合湖泊水体年轻的DOC特征[76 ] ,可明确判断该湖泊br-GDGTs来源于外源陈化土壤的输入。
Prep-HPLC技术被广泛应用于分离纯化环境样品中一类重要的陆源生物标志物——木质素。Ingalls等[39 ] 尝试利用双色谱柱制备液相色谱分离提纯了Pacific Northwest的叶片中的6种木质素酚类化合物。首先,利用反相-HPLC柱(Zorbax Eclipse XDB-C18 column,4.6 mm×150 mm; 5 μm)分离获得半纯的木质素酚类化合物,然后使用正相-HPLC柱(LiChrospher Diol column, 4.6 mm×150 mm, 5 μm)分离和收集纯的木质素酚类化合物单体。因为反相-HPLC不能完全分离所有的木质素酚类化合物,而利用正相-HPLC可以进一步分离在反相-HPLC中会共同溶出的木质素酚类化合物以及与目标木质素酚类化合物具有相似色谱行为的其他基质,从而获得纯度99%以上的目标化合物。相对于双色谱柱-HPLC技术,Hou等[14 ] 利用一个单柱(XDB-C18 )-HPLC技术成功分离了湖泊沉积物中的木质素,该方法在进行HPLC分离之前利用一个C18 -SPE纯化柱去除湖泊沉积物中的中性组分。Feng等[30 ] 通过固相萃取柱结合双色谱柱制备液相色谱分离提纯了华盛顿边缘海沉积物中木质素氧化产物。海洋沉积物尤其是边缘海区域,沉积有机质来源广泛、组成复杂、基质高,因此,相应的木质素酚类化合物分离提纯步骤较为繁琐。首先,通过ENVI-18固相萃取柱将中性化合物等不纯物质去除,然后通过LC-NH2 固相萃取柱将木质素酚类组分为醛/酮类组分和酸性组分。化学纯化后的木质素酚类组分通过Prep-HPLC分离收集用于之后的单体14 C同位素分析。使用Phenomenex Polar-RP柱(4.6 mm×250 mm, 4 μm)进行初步的分离收集,然后使用ZORBAX Eclipse XDB-C18 (4.6 mm×250 mm, 5 μm)进一步的分离纯化。尽管Feng等[30 ] 增加了分离纯化的步骤,但HPLC分离后,目标化合物的回收率(60%~80%)不低于已有报道的回收效率(约为50%)[39 ] ,加之木质素酚类化合物具有高的挥发性,因此导致其在柔和氮气吹干溶剂过程发生较大损失[39 ] 。
Ingalls等[39 ] 评估了Prep-HPLC分离提纯木质素酚类化合物过程中引入的外源碳约有2.2 μg C((1.8±0.9) μg C化石源碳和0.35 μg C现代源碳),接近于其他HPLC分离过程产生的外源碳的贡献[14 ,30 ] 。其中化石源碳的贡献大于80%,主要来自在微波消解Teflon管和/或聚碳酸酯离心管残留中[39 ] 。另外,Feng等[30 ] 利用HPLC和PCGC技术分离的同一样品的Δ14 C结果显示,HPLC分离的香草基酚类化合物的Δ14 C值相比PCGC方法偏负21‰~29‰,二者的差异低于AMS的测试误差。但相较于PCGC方法,HPLC方法避免了大量使用衍生化试剂,且具有进样量大、效率高和低空白的优势。
6 结 语
自从Eglinton等[26 ] 首次将单体分子放射性碳同位素分析技术应用于生物标志物14 C研究以来,该技术发展迅速,越来越多地应用于海洋科学、环境科学、生物地球化学和古气候学等领域,尤其是研究碳源和碳循环有关的问题。并且,随着高灵敏度AMS的快速发展,已经实现了超微样品14 C浓度的测试,甚至于1 μg C的测试[40 ] 。但是,目前CSRA技术发展的最大限制是如何从复杂的自然环境样品中分离和收集纯的、大量的单体分子化合物,同时减少CSRA过程中引入的外源碳的污染[37 ,53 ] 。近些年来,不断改进的PCGC和HPLC技术极大地扩展了生物标志物的应用,同时也提高了目标化合物分离的纯度和回收效率。目前,利用PCGC和Prep-HPLC等制备色谱进行分离和富集生物标志物单体时,对过程空白贡献的外源碳是1~2 μg C,因此,为了确保干扰物质低于10%,至少需要富集10~20 μg C以上的生物标志物单体进行其14 C测试。但据已有研究指出,CSRA技术过程空白具有高度的时空不确定性,所以将来的研究将需要更加关注“零外源碳”的分离纯化过程和严格的质量控制。
The authors have declared that no competing interests exist.
参考文献
文献选项
[1]
Levin I Kromer B Schoch-Fischer H et al. 25 years of tropospheric 14 C observations in central Europe
[J]. Radiocarbon , 1985 , 27 (1 ): 1 -19 .
[本文引用: 1]
[2]
Ardizzone D Cailliet G M Natanson L J et al. Application of bomb radiocarbon chronologies to shortfin mako (Isurus oxyrinchus) age validation
[J]. Environmental Biology of Fishes , 2006 , 77 (3 ): 355 -366 .
[本文引用: 1]
[3]
Zhang Z Zhao M Yang X et al. A hydrocarbon biomarker record for the last 40 kyr of plant input to Lake Heqing, southwestern China
[J]. Organic Geochemistry , 2004 , 35 (5 ): 595 -613 .
[本文引用: 1]
[4]
Wilson A T. Application of AMS 14 C dating to ice core research
[J]. Radiocarbon , 1995 , 37 (2 ): 637 -641 .
[本文引用: 1]
[5]
Pearson A Eglinton T I. The origin of n-alkanes in Santa Monica Basin surface sediment: A model based on compound-specific Δ14 C and δ13 C data
[J]. Organic Geochemistry , 2000 , 31 (11 ): 1 103 -1 116 .
[本文引用: 3]
[6]
Masiello C A Druffel E R M. Black carbon in deep-sea sediments
[J]. Science , 1998 , 280 (5 371 ): 1 911 -1 913 .
[7]
Kusch S Eglinton T I Mix A C et al. Timescales of lateral sediment transport in the Panama Basin as revealed by radiocarbon ages of alkenones, total organic carbon and foraminifera
[J]. Earth and Planetary Science Letters , 2010 , 290 (3/4 ): 340 -350 .
[本文引用: 2]
[8]
Hughen K Lehman S Southon J et al. 14 C activity and global carbon cycle changes over the past 50,000 years
[J]. Science , 2004 , 303 (5 655 ): 202 -207 .
[本文引用: 1]
[9]
Dou Y Yang S Lim D I et al. Provenance discrimination of last deglacial and Holocene sediments in the southwest of Cheju Island, East China Sea
[J]. Palaeogeography, Palaeoclimatology, Palaeoecology , 2015 , 422 : 25 -35 .
[10]
Salgueiro E Naughton F et al . Past circulation along the western Iberian margin: A time slice vision from the Last Glacial to the Holocene
[J]. Quaternary Science Reviews , 2014 , 106 : 316 -329 .
[本文引用: 1]
[11]
Huang Yuanhui Ge Shulan Shi Xuefa et al. An age model reconstruction of Core BR07 from the northern continental slope of the Bering Sea
[J]. Acta Oceanologica Sinica , 2013 , 35 (6 ): 67 -74 .
[本文引用: 1]
[黄元辉 , 葛淑兰 , 石学法 , 等 . 白令海北部陆坡BR07孔年龄框架重建
[J]. 海洋学报 , 2013 , 35 (6 ): 67 -74 .]
[本文引用: 1]
[12]
Kusch S Rethemeyer J Schefu β E et al. Controls on the age of vascular plant biomarkers in Black Sea sediments
[J]. Geochimica et Cosmochimica Acta , 2010 , 74 (24 ): 7 031 -7 047 .
[本文引用: 2]
[13]
Wang Yong Shen Ji Wu Jian et al. Hard-water effect correction of lacustrine sediment ages using the relationship between 14 C levels in lake waters and in the atmosphere: The case of Lake Qinghai
[J]. Journal of Lake Sciences , 2007 , 19 (5 ): 504 -508 .
[本文引用: 1]
[汪勇 , 沈吉 , 吴健 , 等 . 湖泊沉积物14 C年龄硬水效应校正初探——以青海湖为例
[J]. 湖泊科学 , 2007 , 19 (5 ): 504 -508 .]
[本文引用: 1]
[14]
Hou J Huang Y Brodsky C et al. Radiocarbon dating of individual Lignin Phenols: A new approach for establishing chronology of late Quaternary lake sediments
[J]. Analytical Chemistry , 2010 , 82 (17 ): 7 119 -7 126 .
[本文引用: 7]
[15]
Vonk J E Sánchez-García L Semiletov I et al. Molecular and radiocarbon constraints on sources and degradation of terrestrial organic carbon along the Kolyma paleoriver transect, East Siberian Sea
[J]. Biogeosciences , 2010 , 7 (10 ): 3 153 -3 166 .
[本文引用: 1]
[16]
Drenzek N J Montluçon D B Yunker M B et al. Constraints on the origin of sedimentary organic carbon in the Beaufort Sea from coupled molecular 13 C and 14 C measurements
[J]. Marine Chemistry , 2007 , 103 (1/2 ): 146 -162 .
[本文引用: 1]
[17]
Tao S Eglinton T I Montluçon D B et al. Pre-aged soil organic carbon as a major component of the Yellow River suspended load: Regional significance and global relevance
[J]. Earth and Planetary Science Letters , 2015 , 414 : 77 -86 .
[本文引用: 2]
[18]
Tao S Eglinton T I Montluçon D B et al. Diverse origins and pre-depositional histories of organic matter in contemporary Chinese marginal sea sediments
[J]. Geochimica et Cosmochimica Acta , 2016 , 191 : 70 -88 .
[本文引用: 1]
[19]
Matsumoto K Uchida M Kawamura K et al. Radiocarbon variability of fatty acids in semi-urban aerosol samples
[J]. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms , 2004 , 223 (Suppl.C ): 842 -847 .
[20]
Eglinton T I Eglinton G Dupont L et al. Composition, age, and provenance of organic matter in NW African dust over the Atlantic Ocean
[J]. Geochemistry, Geophysics, Geosystems , 2002 , 3 (8 ): 1 -27 .
[本文引用: 1]
[21]
Hedges J I. Global biogeochemical cycles: Progress and problems
[J]. Marine Chemistry , 1992 , 39 (1 ): 67 -93 .
[本文引用: 1]
[22]
Villinski J C Hayes J M Brassell S C et al. Sedimentary sterols as biogeochemical indicators in the Southern Ocean
[J]. Organic Geochemistry , 2008 , 39 (5 ): 567 -588 .
[本文引用: 1]
[23]
Galy V Eglinton T. Protracted storage of biospheric carbon in the Ganges-Brahmaputra Basin
[J]. Nature Geosci , 2011 , 4 (12 ): 843 -847 .
[本文引用: 2]
[24]
Martin E E Ingalls A E Richey J E et al. Age of riverine carbon suggests rapid export of terrestrial primary production in tropics
[J]. Geophysical Research Letters , 2013 , 40 (21 ): 2013 GL057450.
[本文引用: 1]
[25]
Meyers P A. Organic geochemical proxies of paleoceanographic, paleolimnologic, and paleoclimatic processes
[J]. Organic Geochemistry , 1997 , 27 (5/6 ): 213 -250 .
[本文引用: 1]
[26]
Eglinton T I Aluwihare L I Bauer J E et al. Gas chromatographic isolation of individual compounds from complex matrices for radiocarbon dating
[J]. Analytical Chemistry , 1996 , 68 (5 ): 904 -912 .
[本文引用: 14]
[27]
Eglinton T I Bryan C Benitez-Nelson , et al. Variability in radiocarbon ages of individual organic compounds from marine sediments
[J]. Science , 1997 , 277 :796 -799 .
[本文引用: 2]
[28]
Drenzek N J Hughen K A Montluçon D B et al. A new look at old carbon in active margin sediments
[J]. Geology , 2009 , 37 (3 ): 239 -242 .
[本文引用: 1]
[29]
Gustafsson Ö et al . Widespread release of old carbon across the Siberian Arctic echoed by its large rivers
[J]. Biogeosciences , 2011 , 8 (6 ): 1 737 -1 743 .
[30]
Feng X et al . 14 C and 13 C characteristics of higher plant biomarkers in Washington margin surface sediments
[J]. Geochimica et Cosmochimca Acta , 2013 , 105 : 14 -30 .
[本文引用: 10]
[31]
Feng X Vonk J E et al . Differential mobilization of terrestrial carbon pools in Eurasian Arctic river basins
[J]. Proceedings of the National Academy of Sciences , 2013 , 110 (35 ): 14 168 -14 173 .
[本文引用: 1]
[32]
Uchikawa J Brian N P Jane E. et al . Direct application of compound-specific radiocarbon analysis of leaf waxes to establish lacustrine sediment chronology
[J]. Journal of Paleolimnology , 2008 , 39 (1 ): 43 -60 .
[本文引用: 1]
[33]
Matsumoto K Kawamura K Uchida M et al. Compound specific radiocarbon and δ13 C measurements of fatty acids in a continental aerosol sample
[J]. Geophysical Research Letters , 2001 , 28 (24 ): 4 587 -4 590 .
[本文引用: 1]
[34]
Xu L Zheng M Ding X et al. Modern and fossil contributions to polycyclic aromatic hydrocarbons in PM2.5 from north Birmingham, Alabama in the southeastern U.S
[J]. Environmental Science & Technology , 2012 , 46 (3 ): 1 422 -1 429 .
[本文引用: 1]
[35]
Cowie B R Greenberg B M Slater G F. Determination of microbial carbon sources and cycling during remediation of petroleum hydrocarbon impacted soil using natural abundance 14 C analysis of PLFA
[J]. Environmental Science & Technology , 2010 , 44 (7 ): 2 322 -2 327 .
[本文引用: 1]
[36]
Ruff M Wacker L Gäggeler H et al. A gas ion source for radiocarbon measurements at 200 kV
[J]. Radiocarbon , 2007 , 49 (2 ): 307 -314 .
[本文引用: 2]
[37]
Shah S Pearson A. Ultra-microscale (5-25 μg C) analysis of individual lipids by 14 C AMS: Assessment and correction for sample processing blanks
[J]. Radiocarbon , 2007 , 49 (1 ): 69 -82 .
[本文引用: 5]
[38]
Ziolkowski L A Druffel E R M. Quantification of extraneous carbon during compound specific radiocarbon analysis of black carbon
[J]. Analytical Chemistry , 2009 , 81 (24 ): 10 156 -10 161 .
[本文引用: 8]
[39]
Ingalls A E Ellis E E Santos G M et al. HPLC purification of higher plant-dervied lignin phenols for compound specific radiocarbon analysis
[J]. Analytical Chemistry , 2010 , 82 (21 ): 8 931 -8 938 .
[本文引用: 6]
[40]
Santos R A L D , Prange M Castañeda I S et al . Glacial-interglacial variability in Atlantic meridional overturning circulation and thermocline adjustments in the tropical North Atlantic
[J]. Earth and Planetary Science Letters , 2010 , 300 : 407 -414 .
[本文引用: 4]
[41]
Mandalakis M Gustafsson Ö. Optimization of a preparative capillary gas chromatography-mass spectrometry system for the isolation and harvesting of individual polycyclic aromatic hydrocarbons
[J]. Journal of Chromatography A , 2003 , 996 (1/2 ): 163 -172 .
[本文引用: 1]
[42]
Ball G I Xu L Mcnichol A P et al. A two-dimensional, heart-cutting preparative gas chromatograph facilitates highly resolved single-compound isolations with utility towards compound-specific natural abundance radiocarbon (14 C) analyses
[J]. Journal of Chromatography A , 2012 , 1 220 : 122 -131 .
[本文引用: 5]
[43]
Sciarrone D Pantò S Donato P et al. Improving the productivity of a multidimensional chromatographic preparative system by collecting pure chemicals after each of three chromatographic dimensions
[J]. Journal of Chromatography A , 2016 , 1 475 : 80 -85 .
[本文引用: 2]
[44]
Müller P J Kirst G Ruhland G et al. Calibration of the alkenone paleotemperature index based on core-tops from the eastern South Atlantic and the global ocean (60°N~60°S)
[J]. Geochimica et Cosmochimica Acta , 1998 , 62 (10 ): 1 757 -1 772 .
[本文引用: 1]
[45]
Xing L Zhang H Yuan Z et al. Terrestrial and marine biomarker estimates of organic matter sources and distributions in surface sediments from the East China Sea shelf
[J]. Continental Shelf Research , 2011 , 31 (10 ): 1 106 -1 115 .
[本文引用: 1]
[46]
Zhang Z Zhao M Eglinton G et al. Leaf wax lipids as paleovegetational and paleoenvironmental proxies for the Chinese Loess Plateau over the last 170 kyr
[J]. Quaternary Science Reviews , 2006 , 25 (5/6 ): 575 -594 .
[本文引用: 1]
[47]
Calvo E Pelejero C Logan G A. Pressurized liquid extraction of selected molecular biomarkers in deep sea sediments used as proxies in paleoceanography
[J]. Journal of Chromatography A , 2003 , 989 (2 ): 197 -205 .
[本文引用: 1]
[48]
Birkholz A Smittenberg R H Hajdas I et al. Isolation and compound specific radiocarbon dating of terrigenous branched glycerol dialkyl glycerol tetraethers (brGDGTs)
[J]. Organic Geochemistry , 2013 , 60 : 9 -19 .
[本文引用: 4]
[49]
Ohkouchi N Xu L Reddy C M et al. Radiocarbon dating of alkenones from marine sediments: I. Isolation protocol
[J]. Radiocarbon , 2005 , 47 (3 ): 401 -412 .
[本文引用: 4]
[50]
Wacker L Lippold J Molnár M et al. Towards radiocarbon dating of single foraminifera with a gas ion source
[J]. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms , 2013 , 294 (Suppl.C ): 307 -310 .
[本文引用: 1]
[51]
Wacker L Fahrni S M Hajdas I et al. A versatile gas interface for routine radiocarbon analysis with a gas ion source
[J]. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms , 2013 , 294 : 315 -319 .
[本文引用: 1]
[52]
Smith A M Hua Q Williams A et al. Developments in micro-sample 14 C AMS at the ANTARES AMS facility
[J]. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms , 2010 , 268 (7 ): 919 -923 .
[本文引用: 1]
[53]
Mollenhauer G Rethemeyer J. Compound-specific radiocarbon analysis—Analytical challenges and applications
[J]. IOP Conference Series: Earth and Environmental Science , 2009 , 5 (1 ): 1 -9 .
[本文引用: 2]
[54]
Zencak Z Reddy C M Teuten E L et al. Evaluation of gas chromatographic isotope fractionation and process contamination by carbon in compound-specific radiocarbon analysis
[J]. Analytical Chemistry , 2007 , 79 (5 ): 2 042 -2 049 .
[本文引用: 6]
[55]
Buchholz B A Tips and traps in the 14 C bio-AMS preparation laboratory
[J]. Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms , 2000 , 172 (1 ): 404 -408 .
[本文引用: 2]
[56]
Ingalls A E Shah S R Hansman R L et al. Quantifying archaeal community autotrophy in the mesopelagic ocean using natural radiocarbon
[J]. Proceedings of the National Academy of Sciences , 2006 , 103 (17 ): 6 442 -6 447 .
[本文引用: 1]
[57]
Coppola A I Ziolkowski L A Druffel E R M. Extraneous carbon assessments in radiocarbon measurements of black carbon in environmental matrices
[J]. Radiocarbon , 2013 , 55 (3 ): 1 631 -1 640 .
[本文引用: 5]
[58]
Tao Shuqin. The Composition Isotopic Characteristics and Sources of Organic Matter in the Yellow River Suspended Particulates and Adjacent Bohai and Yellow Sea Suface Sediments[D].
Qingdao: Ocean University of China ,2014 .
[本文引用: 1]
[陶舒琴 . 黄河颗粒态及渤、黄海现代沉积有机质的组成和同位素分布特征及源项解析[D]
. 青岛:中国海洋大学 , 2014 .]
[本文引用: 1]
[59]
Santos G M Southon J R Griffin S et al. Ultra small-mass AMS 14 C sample preparation and analyses at KCCAMS/UCI Facility
[J]. Nuclear Instruments and Methods in Physics Research B , 2007 , 259 : 293 -302 .
[本文引用: 1]
[60]
Pearson A. Biogeochemical Applications of Compound-Specific Radiocarbon Analysis[R].
Cambridge: Massachusetts Institute of Technology and Woods Hole Oceanographic Institution , 1999 .
[本文引用: 1]
[61]
Hayes J M Freeman K H Popp B N et al. Compound-specific isotopic analyses: A novel tool for reconstruction of ancient biogeochemical processes
[J]. Organic Geochemistry , 1990 , 16 (4/6 ): 1 115 -1 128 .
[本文引用: 1]
[62]
Caimi R J Brenna J T. Quantitative evaluation of carbon isotopic fractionation during reversed-phase high-performance liquid chromatography
[J]. Journal of Chromatography A , 1997 , 757 (1 ): 307 -310 .
[本文引用: 1]
[63]
Holmstrand H Mandalakis M Zencak Z et al. Chlorine isotope fractionation of a semi-volatile organochlorine compound during preparative megabore-column capillary gas chromatography
[J]. Journal of Chromatography A , 2006 , 1 103 (1 ): 133 -138 .
[本文引用: 1]
[64]
Stuiver M Polach H A. Radiocarbon-discussion reporting of 14 C data
[J]. Radiocarbon , 1977 , 19 (3 ): 355 -363 .
[本文引用: 2]
[65]
Mcnichol A Ertel J Eglinton T I. The radiocarbon content of individual lignin-derived phenols: Technique and initial results
[J]. Radiocarbon , 2000 , 42 (2 ): 219 -227 .
[本文引用: 3]
[66]
Pearson A Mcnichol A P Origins of lipid biomarkers in Santa Monica Basin surface sediment: A case study using compound-specific Δ14 C analysis
[J]. Geochimica et Cosmochimica Acta , 2001 , 65 (18 ): 3 123 -3 137 .
[本文引用: 1]
[67]
Corr L T Berstan R Evershed R P. Optimisation of derivatisation procedures for the determination of δ13 C values of amino acids by gas chromatography/combustion/isotope ratio mass spectrometry
[J]. Rapid Communications in Mass Spectrometry , 2007 , 21 (23 ): 3 759 -3 771 .
[本文引用: 1]
[68]
Shah S R Mollenhauer G Ohkouchi N et al. Origins of archaeal tetraether lipids in sediments: Insights from radiocarbon analysis
[J]. Geochimica et Cosmochimica Acta , 2008 , 72 (18 ): 4 577 -4 594 .
[本文引用: 1]
[69]
Smittenberg R H Hopmans E C Schouten S et al. Rapid isolation of biomarkers for compound specific radiocarbon dating using high-performance liquid chromatography and flow injection analysis-atmospheric pressure chemical ionisation mass spectrometry
[J]. Journal of Chromatography A , 2002 , 978 (1 ): 129 -140 .
[本文引用: 4]
[70]
Bour A L Walker B D Radiocarbon analysis of individual Amino Acids: Carbon blank quantification for a small-sample high-pressure liquid chromatography purification method
[J]. Analytical Chemistry , 2016 , 88 (7 ): 3 521 -3 528 .
[本文引用: 1]
[71]
Hopmans E C et al . A novel proxy for terrestrial organic matter in sediments based on branched and isoprenoid tetraether lipids
[J]. Earth and Planetary Science Letters , 2004 , 224 (1/2 ): 107 -116 .
[本文引用: 1]
[72]
Kim J H Schouten S Buscail R et al. Origin and distribution of terrestrial organic matter in the NW Mediterranean (Gulf of Lions): Exploring the newly developed BIT index
[J]. Geochemistry, Geophysics, Geosystems , 2006 , 7 (11 ),doi:10.1029/2006GC001306 .
[73]
Xing L Zhao M Gao W et al. Multiple proxy estimates of source and spatial variation in organic matter in surface sediments from the southern Yellow Sea
[J]. Organic Geochemistry , 2014 , 76 : 72 -81 .
[本文引用: 1]
[74]
Sinninghe Damsté J S Ossebaar J Abbas B et al . Fluxes and distribution of tetraether lipids in an equatorial African lake: Constraints on the application of the TEX86 palaeothermometer and BIT index in lacustrine settings
[J]. Geochimica et Cosmochimica Acta , 2009 , 73 (14 ): 4 232 -4 249 .
[本文引用: 1]
[75]
Weijers J W H Schefu B E Kim J H et al . Constraints on the sources of branched tetraether membrane lipids in distal marine sediments
[J]. Organic Geochemistry , 2014 , 72 : 14 -22 .
[本文引用: 1]
[76]
Zigah P K Minor E C Werne J P. Radiocarbon and stable-isotope geochemistry of organic and inorganic carbon in Lake Superior
[J]. Global Biogeochemical Cycles , 2012 , 26 (1 ),doi:10.1029/2011GB004132 .
[本文引用: 1]
25 years of tropospheric 14 C observations in central Europe
1
1985
... 天然放射性碳同位素(14 C)主要来源于高层大气层中宇宙射线产生的中子与稳定氮同位素14 N的反应,产生的14 C与大气中的氧结合形成二氧化碳(14 CO2 )进入低层大气层,通过海—气交换和垂直混合进入海洋,成为陆地和海洋生物生命活动(如光合作用和化能自养等的同化作用)的无机碳源.可见,14 C广泛存在于大气、陆地、海洋及生物体中,这使得14 C成为了解生物地球化学碳循环过程和研究各个碳库之间碳交换的有力的示踪工具.另外,人类核试验也是大气层中14 C的重要来源.尤其是20世纪50~60年代密集的核试验,使得北半球大气层的14 C浓度增加了1倍,60年代初期,对流层中CO2 的Δ14 C值超过了900‰[1 ] .自1962年全面禁止核试验以来,大气中由核试验引入的14 CO2 被海洋和陆地生物吸收不断地衰变减少,通过环境生物样品核爆炸标记(“bomb-spike”)的Δ14 C信号的变化,可作为短时间尺度生物地球化学过程的同位素示踪剂[2 ] . ...
Application of bomb radiocarbon chronologies to shortfin mako (Isurus oxyrinchus) age validation
1
2006
... 天然放射性碳同位素(14 C)主要来源于高层大气层中宇宙射线产生的中子与稳定氮同位素14 N的反应,产生的14 C与大气中的氧结合形成二氧化碳(14 CO2 )进入低层大气层,通过海—气交换和垂直混合进入海洋,成为陆地和海洋生物生命活动(如光合作用和化能自养等的同化作用)的无机碳源.可见,14 C广泛存在于大气、陆地、海洋及生物体中,这使得14 C成为了解生物地球化学碳循环过程和研究各个碳库之间碳交换的有力的示踪工具.另外,人类核试验也是大气层中14 C的重要来源.尤其是20世纪50~60年代密集的核试验,使得北半球大气层的14 C浓度增加了1倍,60年代初期,对流层中CO2 的Δ14 C值超过了900‰[1 ] .自1962年全面禁止核试验以来,大气中由核试验引入的14 CO2 被海洋和陆地生物吸收不断地衰变减少,通过环境生物样品核爆炸标记(“bomb-spike”)的Δ14 C信号的变化,可作为短时间尺度生物地球化学过程的同位素示踪剂[2 ] . ...
A hydrocarbon biomarker record for the last 40 kyr of plant input to Lake Heqing, southwestern China
1
2004
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
Application of AMS 14 C dating to ice core research
1
1995
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
The origin of n-alkanes in Santa Monica Basin surface sediment: A model based on compound-specific Δ14 C and δ13 C data
3
2000
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段.Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致.可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标.Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右.但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
Druffel E R M. Black carbon in deep-sea sediments
0
1998
Timescales of lateral sediment transport in the Panama Basin as revealed by radiocarbon ages of alkenones, total organic carbon and foraminifera
2
2010
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
... Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段.Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致.可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标.Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右.但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
14 C activity and global carbon cycle changes over the past 50,000 years
1
2004
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
Provenance discrimination of last deglacial and Holocene sediments in the southwest of Cheju Island, East China Sea
0
2015
Past circulation along the western Iberian margin: A time slice vision from the Last Glacial to the Holocene
1
2014
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
白令海北部陆坡BR07孔年龄框架重建
1
2013
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
白令海北部陆坡BR07孔年龄框架重建
1
2013
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
Controls on the age of vascular plant biomarkers in Black Sea sediments
2
2010
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
湖泊沉积物14 C年龄硬水效应校正初探——以青海湖为例
1
2007
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
湖泊沉积物14 C年龄硬水效应校正初探——以青海湖为例
1
2007
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
Radiocarbon dating of individual Lignin Phenols: A new approach for establishing chronology of late Quaternary lake sediments
7
2010
... 作为传统的定年手段,14 C技术广泛应用于第四纪地质沉积学、古海洋学及古环境学.其原理主要是基于保存在沉积物、土壤或冰芯中总有机碳或无机碳的14 C同位素衰变水平进行时间换算[3 ,4 ] .在第四纪古海洋及古环境研究中,由于受到非海源有机质的输入,总有机质14 C重建的海洋沉积物年龄通常更老一些[5 ~7 ] ,因此通常借助海源生物质定年,消除陆地陈化有机质和古老化石有机碳的影响.有孔虫14 C是应用最为广泛的生物质定年手段[8 ~10 ] .但是,某些海域(比如白令海)钙质生物缺乏,有孔虫等钙质测年材料的缺失制约了古海洋学的研究,从而选择其他生物介质替代指标进行定年.例如硅藻百分含量[11 ] 或海源生物标志物14 C定年[12 ] .在第四纪陆地湖泊古气候环境研究中,湖泊沉积物14 C年龄容易受到湖区搬运的碳酸盐老碳输入的硬水效应影响,导致其年龄偏老[13 ] .Hou等[14 ] 发现,中国青海湖现代湖水溶解无机碳(Dissolved Inorganic Carbon,DIC)的14 C年龄为1 040年,明显低于现代大气CO2 的14 C水平.此外,湖泊的硬水效应在不同历史时期存在较大的差异,所以无法简单地在总有机碳14 C年龄基础上扣除单一恒定的碳库年龄获得准确的沉积物年龄.由于湖相内源有机质需要吸收水体DIC,其14 C年龄受到湖泊硬水效应的影响;而外源的陆生植物(陆相)一般不受这类老碳效应影响.因此,陆地高等植物生物标志物,如木质素的14 C同位素值可被用于陆地湖泊沉积物定年.尤其是类似青海湖这一类形成于碳酸盐基岩之上受硬水效应影响明显的湖泊,陆源生物标志物单体14 C同位素技术提供了一个准确获取沉积物年龄框架的手段[14 ] . ...
... [14 ]. ...
... 自然环境研究中常用生物标志物的含量一般很低(一般小于1 μg/g干重,甚至低于几十个ng/g干重),所以需要通过分离和富集从自然环境样品中获取足够量(>100 μg C)、高纯度的生物标志物以满足AMS的测试要求.为了消除天然环境样品中复杂的基质干扰,首先会利用湿化学法进行目标组分初步的有机溶剂萃取和分离提纯.有机溶剂萃取技术包括超声萃取[44 ,45 ] 、索氏提取[27 ,46 ] 、加速溶剂萃取[47 ,48 ] 以及微波萃取[14 ,17 ] 等技术.在提纯生物标志物过程中,还需要配合一系列的化学分离技术和固相萃取技术等手段.根据化合物的不同化学性质,如中性组分(包括烷烃、醇类和烯酮等)和酸性组分(包括脂肪酸等),可以通过碱水解(0.5 mol/L的KOH的甲醇溶液)将二者分离[49 ] .固相萃取分离技术则是根据不同目标化合物的极性或结构差异实现进一步分离和提纯的化学法.例如,硅胶层析通过改变淋洗液极性梯度,可将不同极性的化合物分离[49 ] ;硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
... Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段.Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致.可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标.Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右.但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
... Birhkolz等[48 ] 改进了已有的Prep-HPLC技术,成功分离富集出湖泊沉积物中br-GDGTs化合物单体.首先利用反相-HPLC(Agilent Eclipse Plus C18 column,4.6 mm×150 mm, 3.5 μm)将br-GDGTs和共流出组分分离;为了除去反相-HPLC柱分离过程中引入的共流出组分和进一步纯化,使用正相-HPLC柱(Alltech Prevail Cyano column, 2.1 mm×150 mm, 3 μm)消除可能来自流动相的外源碳(12 μg C/60 min).改进后的HPLC技术贡献的外源碳为 (0.6±0.2) μg C(其F14 C为0.5±0.2),与Hou等[14 ] 结果基本一致,其外源碳的贡献约为2 μg C(Fm值为0.30).基于br-GDGTs的BIT指标已被广泛用来估算海洋环境中土壤有机质的贡献比例[71 ~73 ] ,但有报道指出湖泊[74 ] 和海洋[75 ] 也会产生br-GDGTs.湖泊、海洋环境样品中br-GDGTs的14 C同位素特征可以为br-GDGTs土壤源指标的应用提供重要的信息.Birkholz等[48 ] 发现Lake Lusvatnet沉积物中的br-GDGTs呈现较老的14 C年龄.结合湖泊水体年轻的DOC特征[76 ] ,可明确判断该湖泊br-GDGTs来源于外源陈化土壤的输入. ...
... Prep-HPLC技术被广泛应用于分离纯化环境样品中一类重要的陆源生物标志物——木质素.Ingalls等[39 ] 尝试利用双色谱柱制备液相色谱分离提纯了Pacific Northwest的叶片中的6种木质素酚类化合物.首先,利用反相-HPLC柱(Zorbax Eclipse XDB-C18 column,4.6 mm×150 mm; 5 μm)分离获得半纯的木质素酚类化合物,然后使用正相-HPLC柱(LiChrospher Diol column, 4.6 mm×150 mm, 5 μm)分离和收集纯的木质素酚类化合物单体.因为反相-HPLC不能完全分离所有的木质素酚类化合物,而利用正相-HPLC可以进一步分离在反相-HPLC中会共同溶出的木质素酚类化合物以及与目标木质素酚类化合物具有相似色谱行为的其他基质,从而获得纯度99%以上的目标化合物.相对于双色谱柱-HPLC技术,Hou等[14 ] 利用一个单柱(XDB-C18 )-HPLC技术成功分离了湖泊沉积物中的木质素,该方法在进行HPLC分离之前利用一个C18 -SPE纯化柱去除湖泊沉积物中的中性组分.Feng等[30 ] 通过固相萃取柱结合双色谱柱制备液相色谱分离提纯了华盛顿边缘海沉积物中木质素氧化产物.海洋沉积物尤其是边缘海区域,沉积有机质来源广泛、组成复杂、基质高,因此,相应的木质素酚类化合物分离提纯步骤较为繁琐.首先,通过ENVI-18固相萃取柱将中性化合物等不纯物质去除,然后通过LC-NH2 固相萃取柱将木质素酚类组分为醛/酮类组分和酸性组分.化学纯化后的木质素酚类组分通过Prep-HPLC分离收集用于之后的单体14 C同位素分析.使用Phenomenex Polar-RP柱(4.6 mm×250 mm, 4 μm)进行初步的分离收集,然后使用ZORBAX Eclipse XDB-C18 (4.6 mm×250 mm, 5 μm)进一步的分离纯化.尽管Feng等[30 ] 增加了分离纯化的步骤,但HPLC分离后,目标化合物的回收率(60%~80%)不低于已有报道的回收效率(约为50%)[39 ] ,加之木质素酚类化合物具有高的挥发性,因此导致其在柔和氮气吹干溶剂过程发生较大损失[39 ] . ...
... Ingalls等[39 ] 评估了Prep-HPLC分离提纯木质素酚类化合物过程中引入的外源碳约有2.2 μg C((1.8±0.9) μg C化石源碳和0.35 μg C现代源碳),接近于其他HPLC分离过程产生的外源碳的贡献[14 ,30 ] .其中化石源碳的贡献大于80%,主要来自在微波消解Teflon管和/或聚碳酸酯离心管残留中[39 ] .另外,Feng等[30 ] 利用HPLC和PCGC技术分离的同一样品的Δ14 C结果显示,HPLC分离的香草基酚类化合物的Δ14 C值相比PCGC方法偏负21‰~29‰,二者的差异低于AMS的测试误差.但相较于PCGC方法,HPLC方法避免了大量使用衍生化试剂,且具有进样量大、效率高和低空白的优势. ...
Molecular and radiocarbon constraints on sources and degradation of terrestrial organic carbon along the Kolyma paleoriver transect, East Siberian Sea
1
2010
... 另外,生物标志物单体14 C同位素也是一类常见的来源示踪指标,被广泛用于大气颗粒物、土壤、河流、湖泊、海洋等生态环境系统中有机碳的来源解析研究中[15 ~20 ] .由于高的初级生产力以及高的陆源输入,边缘海不仅是重要的海洋碳汇,也是一个重要的陆源碳汇.有机碳储库(carbon pools)(颗粒有机碳汇和溶解有机碳汇)中不同来源有机质的比例不仅决定了其组成性质和埋藏效率,而且还在不同尺度上影响全球碳循环.通过生物标志物单体14 C同位素技术可以将自然环境系统中有机质来源从“年龄”的角度基本可划分为三大类,包括年轻的海源有机质和陆源维管植物残留物(Δ14 C=0±50‰,近现代: <100年)、陈化的土壤有机质(千年尺度)及古老的沉积岩风化有机质(Δ14 C=-1 000‰,万年尺度)[21 ,22 ] .以上3种有机质中只有海洋生源和植被源有机碳汇能够影响短时间尺度大气CO2 的浓度,而陈化土壤源和化石源古老有机质常常会在陆地湖泊、土壤或沉积岩等中间有机碳储库中埋藏成百上千年甚至百万年,随时间的推移,通过河流或者风尘等传输过程进入海洋沉积环境中,所以这些陆源有机质的埋藏在短时间尺度上对近现代大气CO2 的埋藏贡献几乎为零.因此,通过14 C同位素特征将现代有机碳和其他年龄的有机碳区分开,有助于正确评估现代海洋“碳汇”的功能,并从“年龄”的新视角认识有机碳的迁移、转化和埋藏特征.大部分自然样品中有机碳的来源复杂,这一异质性导致总有机质的表观14 C同位素组成代表的是一个混合的信号.尤其是陈化土壤有机质的Δ14 C端元值的高度时空可变性[23 ,24 ] ,很难通过在自然界中找到对应的典型样品来确定它,导致总有机质14 C同位素信号无法区分和剥离较老的土壤源有机质和古老的化石源有机质的信号.由于有机物的14 C同位素特征不依赖于生物合成方式和化合物种类,只受合成时利用的碳源的14 C同位素水平和“中间储库”停留时间控制.生物标志物由特定生物合成,性质稳定,在分子水平上指示其特定来源[25 ] ,生物标志物14 C同位素组成被广泛应用于评价生物圈中有机质的来源及组成变化[16 ,26 ~31 ] .因此,通过来源具有专一性(Source Specific Characteristics)不同生物标志物单体14 C信号可以准确获取相应来源有机质的14 C同位素特征端元值,以达到示踪这类有机物“源—汇”过程变化. ...
Constraints on the origin of sedimentary organic carbon in the Beaufort Sea from coupled molecular 13 C and 14 C measurements
1
2007
... 另外,生物标志物单体14 C同位素也是一类常见的来源示踪指标,被广泛用于大气颗粒物、土壤、河流、湖泊、海洋等生态环境系统中有机碳的来源解析研究中[15 ~20 ] .由于高的初级生产力以及高的陆源输入,边缘海不仅是重要的海洋碳汇,也是一个重要的陆源碳汇.有机碳储库(carbon pools)(颗粒有机碳汇和溶解有机碳汇)中不同来源有机质的比例不仅决定了其组成性质和埋藏效率,而且还在不同尺度上影响全球碳循环.通过生物标志物单体14 C同位素技术可以将自然环境系统中有机质来源从“年龄”的角度基本可划分为三大类,包括年轻的海源有机质和陆源维管植物残留物(Δ14 C=0±50‰,近现代: <100年)、陈化的土壤有机质(千年尺度)及古老的沉积岩风化有机质(Δ14 C=-1 000‰,万年尺度)[21 ,22 ] .以上3种有机质中只有海洋生源和植被源有机碳汇能够影响短时间尺度大气CO2 的浓度,而陈化土壤源和化石源古老有机质常常会在陆地湖泊、土壤或沉积岩等中间有机碳储库中埋藏成百上千年甚至百万年,随时间的推移,通过河流或者风尘等传输过程进入海洋沉积环境中,所以这些陆源有机质的埋藏在短时间尺度上对近现代大气CO2 的埋藏贡献几乎为零.因此,通过14 C同位素特征将现代有机碳和其他年龄的有机碳区分开,有助于正确评估现代海洋“碳汇”的功能,并从“年龄”的新视角认识有机碳的迁移、转化和埋藏特征.大部分自然样品中有机碳的来源复杂,这一异质性导致总有机质的表观14 C同位素组成代表的是一个混合的信号.尤其是陈化土壤有机质的Δ14 C端元值的高度时空可变性[23 ,24 ] ,很难通过在自然界中找到对应的典型样品来确定它,导致总有机质14 C同位素信号无法区分和剥离较老的土壤源有机质和古老的化石源有机质的信号.由于有机物的14 C同位素特征不依赖于生物合成方式和化合物种类,只受合成时利用的碳源的14 C同位素水平和“中间储库”停留时间控制.生物标志物由特定生物合成,性质稳定,在分子水平上指示其特定来源[25 ] ,生物标志物14 C同位素组成被广泛应用于评价生物圈中有机质的来源及组成变化[16 ,26 ~31 ] .因此,通过来源具有专一性(Source Specific Characteristics)不同生物标志物单体14 C信号可以准确获取相应来源有机质的14 C同位素特征端元值,以达到示踪这类有机物“源—汇”过程变化. ...
Pre-aged soil organic carbon as a major component of the Yellow River suspended load: Regional significance and global relevance
2
2015
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... 自然环境研究中常用生物标志物的含量一般很低(一般小于1 μg/g干重,甚至低于几十个ng/g干重),所以需要通过分离和富集从自然环境样品中获取足够量(>100 μg C)、高纯度的生物标志物以满足AMS的测试要求.为了消除天然环境样品中复杂的基质干扰,首先会利用湿化学法进行目标组分初步的有机溶剂萃取和分离提纯.有机溶剂萃取技术包括超声萃取[44 ,45 ] 、索氏提取[27 ,46 ] 、加速溶剂萃取[47 ,48 ] 以及微波萃取[14 ,17 ] 等技术.在提纯生物标志物过程中,还需要配合一系列的化学分离技术和固相萃取技术等手段.根据化合物的不同化学性质,如中性组分(包括烷烃、醇类和烯酮等)和酸性组分(包括脂肪酸等),可以通过碱水解(0.5 mol/L的KOH的甲醇溶液)将二者分离[49 ] .固相萃取分离技术则是根据不同目标化合物的极性或结构差异实现进一步分离和提纯的化学法.例如,硅胶层析通过改变淋洗液极性梯度,可将不同极性的化合物分离[49 ] ;硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
Diverse origins and pre-depositional histories of organic matter in contemporary Chinese marginal sea sediments
1
2016
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
Radiocarbon variability of fatty acids in semi-urban aerosol samples
0
2004
Composition, age, and provenance of organic matter in NW African dust over the Atlantic Ocean
1
2002
... 另外,生物标志物单体14 C同位素也是一类常见的来源示踪指标,被广泛用于大气颗粒物、土壤、河流、湖泊、海洋等生态环境系统中有机碳的来源解析研究中[15 ~20 ] .由于高的初级生产力以及高的陆源输入,边缘海不仅是重要的海洋碳汇,也是一个重要的陆源碳汇.有机碳储库(carbon pools)(颗粒有机碳汇和溶解有机碳汇)中不同来源有机质的比例不仅决定了其组成性质和埋藏效率,而且还在不同尺度上影响全球碳循环.通过生物标志物单体14 C同位素技术可以将自然环境系统中有机质来源从“年龄”的角度基本可划分为三大类,包括年轻的海源有机质和陆源维管植物残留物(Δ14 C=0±50‰,近现代: <100年)、陈化的土壤有机质(千年尺度)及古老的沉积岩风化有机质(Δ14 C=-1 000‰,万年尺度)[21 ,22 ] .以上3种有机质中只有海洋生源和植被源有机碳汇能够影响短时间尺度大气CO2 的浓度,而陈化土壤源和化石源古老有机质常常会在陆地湖泊、土壤或沉积岩等中间有机碳储库中埋藏成百上千年甚至百万年,随时间的推移,通过河流或者风尘等传输过程进入海洋沉积环境中,所以这些陆源有机质的埋藏在短时间尺度上对近现代大气CO2 的埋藏贡献几乎为零.因此,通过14 C同位素特征将现代有机碳和其他年龄的有机碳区分开,有助于正确评估现代海洋“碳汇”的功能,并从“年龄”的新视角认识有机碳的迁移、转化和埋藏特征.大部分自然样品中有机碳的来源复杂,这一异质性导致总有机质的表观14 C同位素组成代表的是一个混合的信号.尤其是陈化土壤有机质的Δ14 C端元值的高度时空可变性[23 ,24 ] ,很难通过在自然界中找到对应的典型样品来确定它,导致总有机质14 C同位素信号无法区分和剥离较老的土壤源有机质和古老的化石源有机质的信号.由于有机物的14 C同位素特征不依赖于生物合成方式和化合物种类,只受合成时利用的碳源的14 C同位素水平和“中间储库”停留时间控制.生物标志物由特定生物合成,性质稳定,在分子水平上指示其特定来源[25 ] ,生物标志物14 C同位素组成被广泛应用于评价生物圈中有机质的来源及组成变化[16 ,26 ~31 ] .因此,通过来源具有专一性(Source Specific Characteristics)不同生物标志物单体14 C信号可以准确获取相应来源有机质的14 C同位素特征端元值,以达到示踪这类有机物“源—汇”过程变化. ...
Global biogeochemical cycles: Progress and problems
1
1992
... 另外,生物标志物单体14 C同位素也是一类常见的来源示踪指标,被广泛用于大气颗粒物、土壤、河流、湖泊、海洋等生态环境系统中有机碳的来源解析研究中[15 ~20 ] .由于高的初级生产力以及高的陆源输入,边缘海不仅是重要的海洋碳汇,也是一个重要的陆源碳汇.有机碳储库(carbon pools)(颗粒有机碳汇和溶解有机碳汇)中不同来源有机质的比例不仅决定了其组成性质和埋藏效率,而且还在不同尺度上影响全球碳循环.通过生物标志物单体14 C同位素技术可以将自然环境系统中有机质来源从“年龄”的角度基本可划分为三大类,包括年轻的海源有机质和陆源维管植物残留物(Δ14 C=0±50‰,近现代: <100年)、陈化的土壤有机质(千年尺度)及古老的沉积岩风化有机质(Δ14 C=-1 000‰,万年尺度)[21 ,22 ] .以上3种有机质中只有海洋生源和植被源有机碳汇能够影响短时间尺度大气CO2 的浓度,而陈化土壤源和化石源古老有机质常常会在陆地湖泊、土壤或沉积岩等中间有机碳储库中埋藏成百上千年甚至百万年,随时间的推移,通过河流或者风尘等传输过程进入海洋沉积环境中,所以这些陆源有机质的埋藏在短时间尺度上对近现代大气CO2 的埋藏贡献几乎为零.因此,通过14 C同位素特征将现代有机碳和其他年龄的有机碳区分开,有助于正确评估现代海洋“碳汇”的功能,并从“年龄”的新视角认识有机碳的迁移、转化和埋藏特征.大部分自然样品中有机碳的来源复杂,这一异质性导致总有机质的表观14 C同位素组成代表的是一个混合的信号.尤其是陈化土壤有机质的Δ14 C端元值的高度时空可变性[23 ,24 ] ,很难通过在自然界中找到对应的典型样品来确定它,导致总有机质14 C同位素信号无法区分和剥离较老的土壤源有机质和古老的化石源有机质的信号.由于有机物的14 C同位素特征不依赖于生物合成方式和化合物种类,只受合成时利用的碳源的14 C同位素水平和“中间储库”停留时间控制.生物标志物由特定生物合成,性质稳定,在分子水平上指示其特定来源[25 ] ,生物标志物14 C同位素组成被广泛应用于评价生物圈中有机质的来源及组成变化[16 ,26 ~31 ] .因此,通过来源具有专一性(Source Specific Characteristics)不同生物标志物单体14 C信号可以准确获取相应来源有机质的14 C同位素特征端元值,以达到示踪这类有机物“源—汇”过程变化. ...
Sedimentary sterols as biogeochemical indicators in the Southern Ocean
1
2008
... 另外,生物标志物单体14 C同位素也是一类常见的来源示踪指标,被广泛用于大气颗粒物、土壤、河流、湖泊、海洋等生态环境系统中有机碳的来源解析研究中[15 ~20 ] .由于高的初级生产力以及高的陆源输入,边缘海不仅是重要的海洋碳汇,也是一个重要的陆源碳汇.有机碳储库(carbon pools)(颗粒有机碳汇和溶解有机碳汇)中不同来源有机质的比例不仅决定了其组成性质和埋藏效率,而且还在不同尺度上影响全球碳循环.通过生物标志物单体14 C同位素技术可以将自然环境系统中有机质来源从“年龄”的角度基本可划分为三大类,包括年轻的海源有机质和陆源维管植物残留物(Δ14 C=0±50‰,近现代: <100年)、陈化的土壤有机质(千年尺度)及古老的沉积岩风化有机质(Δ14 C=-1 000‰,万年尺度)[21 ,22 ] .以上3种有机质中只有海洋生源和植被源有机碳汇能够影响短时间尺度大气CO2 的浓度,而陈化土壤源和化石源古老有机质常常会在陆地湖泊、土壤或沉积岩等中间有机碳储库中埋藏成百上千年甚至百万年,随时间的推移,通过河流或者风尘等传输过程进入海洋沉积环境中,所以这些陆源有机质的埋藏在短时间尺度上对近现代大气CO2 的埋藏贡献几乎为零.因此,通过14 C同位素特征将现代有机碳和其他年龄的有机碳区分开,有助于正确评估现代海洋“碳汇”的功能,并从“年龄”的新视角认识有机碳的迁移、转化和埋藏特征.大部分自然样品中有机碳的来源复杂,这一异质性导致总有机质的表观14 C同位素组成代表的是一个混合的信号.尤其是陈化土壤有机质的Δ14 C端元值的高度时空可变性[23 ,24 ] ,很难通过在自然界中找到对应的典型样品来确定它,导致总有机质14 C同位素信号无法区分和剥离较老的土壤源有机质和古老的化石源有机质的信号.由于有机物的14 C同位素特征不依赖于生物合成方式和化合物种类,只受合成时利用的碳源的14 C同位素水平和“中间储库”停留时间控制.生物标志物由特定生物合成,性质稳定,在分子水平上指示其特定来源[25 ] ,生物标志物14 C同位素组成被广泛应用于评价生物圈中有机质的来源及组成变化[16 ,26 ~31 ] .因此,通过来源具有专一性(Source Specific Characteristics)不同生物标志物单体14 C信号可以准确获取相应来源有机质的14 C同位素特征端元值,以达到示踪这类有机物“源—汇”过程变化. ...
Protracted storage of biospheric carbon in the Ganges-Brahmaputra Basin
2
2011
... 另外,生物标志物单体14 C同位素也是一类常见的来源示踪指标,被广泛用于大气颗粒物、土壤、河流、湖泊、海洋等生态环境系统中有机碳的来源解析研究中[15 ~20 ] .由于高的初级生产力以及高的陆源输入,边缘海不仅是重要的海洋碳汇,也是一个重要的陆源碳汇.有机碳储库(carbon pools)(颗粒有机碳汇和溶解有机碳汇)中不同来源有机质的比例不仅决定了其组成性质和埋藏效率,而且还在不同尺度上影响全球碳循环.通过生物标志物单体14 C同位素技术可以将自然环境系统中有机质来源从“年龄”的角度基本可划分为三大类,包括年轻的海源有机质和陆源维管植物残留物(Δ14 C=0±50‰,近现代: <100年)、陈化的土壤有机质(千年尺度)及古老的沉积岩风化有机质(Δ14 C=-1 000‰,万年尺度)[21 ,22 ] .以上3种有机质中只有海洋生源和植被源有机碳汇能够影响短时间尺度大气CO2 的浓度,而陈化土壤源和化石源古老有机质常常会在陆地湖泊、土壤或沉积岩等中间有机碳储库中埋藏成百上千年甚至百万年,随时间的推移,通过河流或者风尘等传输过程进入海洋沉积环境中,所以这些陆源有机质的埋藏在短时间尺度上对近现代大气CO2 的埋藏贡献几乎为零.因此,通过14 C同位素特征将现代有机碳和其他年龄的有机碳区分开,有助于正确评估现代海洋“碳汇”的功能,并从“年龄”的新视角认识有机碳的迁移、转化和埋藏特征.大部分自然样品中有机碳的来源复杂,这一异质性导致总有机质的表观14 C同位素组成代表的是一个混合的信号.尤其是陈化土壤有机质的Δ14 C端元值的高度时空可变性[23 ,24 ] ,很难通过在自然界中找到对应的典型样品来确定它,导致总有机质14 C同位素信号无法区分和剥离较老的土壤源有机质和古老的化石源有机质的信号.由于有机物的14 C同位素特征不依赖于生物合成方式和化合物种类,只受合成时利用的碳源的14 C同位素水平和“中间储库”停留时间控制.生物标志物由特定生物合成,性质稳定,在分子水平上指示其特定来源[25 ] ,生物标志物14 C同位素组成被广泛应用于评价生物圈中有机质的来源及组成变化[16 ,26 ~31 ] .因此,通过来源具有专一性(Source Specific Characteristics)不同生物标志物单体14 C信号可以准确获取相应来源有机质的14 C同位素特征端元值,以达到示踪这类有机物“源—汇”过程变化. ...
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
Age of riverine carbon suggests rapid export of terrestrial primary production in tropics
1
2013
... 另外,生物标志物单体14 C同位素也是一类常见的来源示踪指标,被广泛用于大气颗粒物、土壤、河流、湖泊、海洋等生态环境系统中有机碳的来源解析研究中[15 ~20 ] .由于高的初级生产力以及高的陆源输入,边缘海不仅是重要的海洋碳汇,也是一个重要的陆源碳汇.有机碳储库(carbon pools)(颗粒有机碳汇和溶解有机碳汇)中不同来源有机质的比例不仅决定了其组成性质和埋藏效率,而且还在不同尺度上影响全球碳循环.通过生物标志物单体14 C同位素技术可以将自然环境系统中有机质来源从“年龄”的角度基本可划分为三大类,包括年轻的海源有机质和陆源维管植物残留物(Δ14 C=0±50‰,近现代: <100年)、陈化的土壤有机质(千年尺度)及古老的沉积岩风化有机质(Δ14 C=-1 000‰,万年尺度)[21 ,22 ] .以上3种有机质中只有海洋生源和植被源有机碳汇能够影响短时间尺度大气CO2 的浓度,而陈化土壤源和化石源古老有机质常常会在陆地湖泊、土壤或沉积岩等中间有机碳储库中埋藏成百上千年甚至百万年,随时间的推移,通过河流或者风尘等传输过程进入海洋沉积环境中,所以这些陆源有机质的埋藏在短时间尺度上对近现代大气CO2 的埋藏贡献几乎为零.因此,通过14 C同位素特征将现代有机碳和其他年龄的有机碳区分开,有助于正确评估现代海洋“碳汇”的功能,并从“年龄”的新视角认识有机碳的迁移、转化和埋藏特征.大部分自然样品中有机碳的来源复杂,这一异质性导致总有机质的表观14 C同位素组成代表的是一个混合的信号.尤其是陈化土壤有机质的Δ14 C端元值的高度时空可变性[23 ,24 ] ,很难通过在自然界中找到对应的典型样品来确定它,导致总有机质14 C同位素信号无法区分和剥离较老的土壤源有机质和古老的化石源有机质的信号.由于有机物的14 C同位素特征不依赖于生物合成方式和化合物种类,只受合成时利用的碳源的14 C同位素水平和“中间储库”停留时间控制.生物标志物由特定生物合成,性质稳定,在分子水平上指示其特定来源[25 ] ,生物标志物14 C同位素组成被广泛应用于评价生物圈中有机质的来源及组成变化[16 ,26 ~31 ] .因此,通过来源具有专一性(Source Specific Characteristics)不同生物标志物单体14 C信号可以准确获取相应来源有机质的14 C同位素特征端元值,以达到示踪这类有机物“源—汇”过程变化. ...
Organic geochemical proxies of paleoceanographic, paleolimnologic, and paleoclimatic processes
1
1997
... 另外,生物标志物单体14 C同位素也是一类常见的来源示踪指标,被广泛用于大气颗粒物、土壤、河流、湖泊、海洋等生态环境系统中有机碳的来源解析研究中[15 ~20 ] .由于高的初级生产力以及高的陆源输入,边缘海不仅是重要的海洋碳汇,也是一个重要的陆源碳汇.有机碳储库(carbon pools)(颗粒有机碳汇和溶解有机碳汇)中不同来源有机质的比例不仅决定了其组成性质和埋藏效率,而且还在不同尺度上影响全球碳循环.通过生物标志物单体14 C同位素技术可以将自然环境系统中有机质来源从“年龄”的角度基本可划分为三大类,包括年轻的海源有机质和陆源维管植物残留物(Δ14 C=0±50‰,近现代: <100年)、陈化的土壤有机质(千年尺度)及古老的沉积岩风化有机质(Δ14 C=-1 000‰,万年尺度)[21 ,22 ] .以上3种有机质中只有海洋生源和植被源有机碳汇能够影响短时间尺度大气CO2 的浓度,而陈化土壤源和化石源古老有机质常常会在陆地湖泊、土壤或沉积岩等中间有机碳储库中埋藏成百上千年甚至百万年,随时间的推移,通过河流或者风尘等传输过程进入海洋沉积环境中,所以这些陆源有机质的埋藏在短时间尺度上对近现代大气CO2 的埋藏贡献几乎为零.因此,通过14 C同位素特征将现代有机碳和其他年龄的有机碳区分开,有助于正确评估现代海洋“碳汇”的功能,并从“年龄”的新视角认识有机碳的迁移、转化和埋藏特征.大部分自然样品中有机碳的来源复杂,这一异质性导致总有机质的表观14 C同位素组成代表的是一个混合的信号.尤其是陈化土壤有机质的Δ14 C端元值的高度时空可变性[23 ,24 ] ,很难通过在自然界中找到对应的典型样品来确定它,导致总有机质14 C同位素信号无法区分和剥离较老的土壤源有机质和古老的化石源有机质的信号.由于有机物的14 C同位素特征不依赖于生物合成方式和化合物种类,只受合成时利用的碳源的14 C同位素水平和“中间储库”停留时间控制.生物标志物由特定生物合成,性质稳定,在分子水平上指示其特定来源[25 ] ,生物标志物14 C同位素组成被广泛应用于评价生物圈中有机质的来源及组成变化[16 ,26 ~31 ] .因此,通过来源具有专一性(Source Specific Characteristics)不同生物标志物单体14 C信号可以准确获取相应来源有机质的14 C同位素特征端元值,以达到示踪这类有机物“源—汇”过程变化. ...
Gas chromatographic isolation of individual compounds from complex matrices for radiocarbon dating
14
1996
... 另外,生物标志物单体14 C同位素也是一类常见的来源示踪指标,被广泛用于大气颗粒物、土壤、河流、湖泊、海洋等生态环境系统中有机碳的来源解析研究中[15 ~20 ] .由于高的初级生产力以及高的陆源输入,边缘海不仅是重要的海洋碳汇,也是一个重要的陆源碳汇.有机碳储库(carbon pools)(颗粒有机碳汇和溶解有机碳汇)中不同来源有机质的比例不仅决定了其组成性质和埋藏效率,而且还在不同尺度上影响全球碳循环.通过生物标志物单体14 C同位素技术可以将自然环境系统中有机质来源从“年龄”的角度基本可划分为三大类,包括年轻的海源有机质和陆源维管植物残留物(Δ14 C=0±50‰,近现代: <100年)、陈化的土壤有机质(千年尺度)及古老的沉积岩风化有机质(Δ14 C=-1 000‰,万年尺度)[21 ,22 ] .以上3种有机质中只有海洋生源和植被源有机碳汇能够影响短时间尺度大气CO2 的浓度,而陈化土壤源和化石源古老有机质常常会在陆地湖泊、土壤或沉积岩等中间有机碳储库中埋藏成百上千年甚至百万年,随时间的推移,通过河流或者风尘等传输过程进入海洋沉积环境中,所以这些陆源有机质的埋藏在短时间尺度上对近现代大气CO2 的埋藏贡献几乎为零.因此,通过14 C同位素特征将现代有机碳和其他年龄的有机碳区分开,有助于正确评估现代海洋“碳汇”的功能,并从“年龄”的新视角认识有机碳的迁移、转化和埋藏特征.大部分自然样品中有机碳的来源复杂,这一异质性导致总有机质的表观14 C同位素组成代表的是一个混合的信号.尤其是陈化土壤有机质的Δ14 C端元值的高度时空可变性[23 ,24 ] ,很难通过在自然界中找到对应的典型样品来确定它,导致总有机质14 C同位素信号无法区分和剥离较老的土壤源有机质和古老的化石源有机质的信号.由于有机物的14 C同位素特征不依赖于生物合成方式和化合物种类,只受合成时利用的碳源的14 C同位素水平和“中间储库”停留时间控制.生物标志物由特定生物合成,性质稳定,在分子水平上指示其特定来源[25 ] ,生物标志物14 C同位素组成被广泛应用于评价生物圈中有机质的来源及组成变化[16 ,26 ~31 ] .因此,通过来源具有专一性(Source Specific Characteristics)不同生物标志物单体14 C信号可以准确获取相应来源有机质的14 C同位素特征端元值,以达到示踪这类有机物“源—汇”过程变化. ...
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... [26 ]成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... 天然样品的CSRA分析中,PCGC是最早发展起来的用于同位素分析的高纯度单体制备色谱技术.PCGC具有高的分离能力,克服了毛细管气相色谱的有限容量(<500 ng/compound,compound为单个化合物)的技术限制.整个装置由2部分组成:气相色谱和Gerstel八通阀回收装置,共6个回收窗口(图1 ).气相色谱端耦合了CIS系统程序升温的PTV进样口、大口径色谱柱(0.53 mm(内径,i, d),0.5 μm(固定相膜厚,film thickness))、FID检测器、十字分流阀.溶剂排空方式的PTV进样口和大孔径色谱柱的使用可实现大体积(≥5 μL/injection)、大容量(约2 μg/compound)进样,通过30~50次的重复进样可以收集足够量的目标化合物(60~100 μg/compound)满足现代高灵敏度AMS[50 ,51 ] 的14 C同位素测定需要.经过高效气相色谱柱分离的目标有机化合物单体通过十字分流阀,约1%进入FID检测器,其余99%通过毛细管传输线(transfer line,0.32 mm(内径,i.d.))进入Gerstel八通阀回收装置;低的保留时间漂移程度(标准偏差最大为0.03 min)保证目标化合物多次收集的准确性和可靠性[26 ] .Eglinton等[26 ] 首次应用PCGC分离富集了海洋沉积物中常见的类脂生物标志物单体,实现了自然环境浓度水平的单体14 C测定.然而CSRA技术仍然遇到很多的挑战和限制,导致其还没有真正实现全自动化,甚至相关的质量控制标准也未建立.在自然环境研究中,CSRA技术的应用所面临的挑战和瓶颈主要是环境样品中常见的几类生物标志物含量都较低,其有机化合物单体的纯化制备过程复杂繁琐且技术要求高.随着高灵敏度加速器质谱的发展,14 C同位素测定所需的样品量越来越小,甚至能够测试最低至几个微克碳的样品[36 ,52 ] .因此,过程空白或外源碳(Cex )对样品尤其是小样品最终14 C结果产生的影响就愈加明显[37 ,40 ,53 ] . ...
... [26 ]首次应用PCGC分离富集了海洋沉积物中常见的类脂生物标志物单体,实现了自然环境浓度水平的单体14 C测定.然而CSRA技术仍然遇到很多的挑战和限制,导致其还没有真正实现全自动化,甚至相关的质量控制标准也未建立.在自然环境研究中,CSRA技术的应用所面临的挑战和瓶颈主要是环境样品中常见的几类生物标志物含量都较低,其有机化合物单体的纯化制备过程复杂繁琐且技术要求高.随着高灵敏度加速器质谱的发展,14 C同位素测定所需的样品量越来越小,甚至能够测试最低至几个微克碳的样品[36 ,52 ] .因此,过程空白或外源碳(Cex )对样品尤其是小样品最终14 C结果产生的影响就愈加明显[37 ,40 ,53 ] . ...
... PCGC系统工作原理图示[26 ] ...
... Diagrammatic representation of the PCGC instrument[26 ] ...
... 随着CSRA技术在自然环境样品中的应用,样品量逐渐向超微样品(<25 μg C)发展,此时过程空白也就成为了CSRA样品14 C测试精确度和准确度的实际限制因素[37 ,56 ] .自然环境中的样品,化学萃取和PCGC分离富集过程中引入的外源碳是过程空白的主要贡献,远远高于在真空线转换为CO2 和/或石墨化过程中外源碳的量;并且当样品量变小时(<100 μg C),过程空白的评价显得更为重要[40 ] .在PCGC分离过程中,色谱柱流失是外源碳的主要来源之一.Eglinton等[26 ] 发现,当PCGC使用高容量色谱柱(>1 μm固定相膜厚)时,尤其是高沸点的有机化合物单体组分中,GC-FID能够明显从PCGC-U型捕集管中检测出聚甲基硅酮的降解产物;而对于使用较薄的固定相时(≤0.5 μm),色谱柱流失的影响则明显减少. ...
... 定量估算PCGC分离过程所引入外源碳的方法分别为直接法(directly method)和间接法(indirectly method)[38 ,57 ] .直接法是假定在PCGC分离过程中的外源碳主要是来自柱流失(包括过去样品的残留和/或GC色谱柱固定相的脱落),且柱流失不随时间和温度的变化而改变.在无溶剂条件下运行PCGC,馏分收集器按照目标化合物的截留时间收集相同条件下的仪器本底空白.Ziolkowski等[38 ] 通过直接法评估了PCGC分离过程中空白的质量随馏分收集器的收集窗口时间增加而增加,平均产生外源碳为(0.1±0.05) μg C /min(Fm=0.125±0.034).而间接方法则是通过加入已知14 C同位素水平的参考标准——“现代源(modern)”有机化合物标准(Fm=1)和“化石源(fossil)”有机化合物标准(Fm=0),分别评估PCGC分离过程中引入的化石源空白碳和现代空白碳的质量.二者质量加和即为引入的外源碳空白总质量,二者Fm的质量加权平均值即为外源碳空白的同位素特征值[38 ] .Ziolkowski等[38 ] 的研究结果显示通过间接法评估得到的PCGC分离过程中引入的外源碳中化石源空白碳和现代源空白碳的贡献分别是Cpcgc-fossil =(0.4±0.2) μg C/min和Cpcgc-modern =(0.2±0.1) μg C/min.因此,应用间接方法估算的外源碳的贡献是Cpcgc =(0.6±0.3) μg C/min(Fm=0.3±0.1).2种方法估算的PCGC分离过程中引入外源碳的贡献相差较大(0.5 μg C),这可能是在有溶剂条件下,来自色谱柱的外源碳更容易被洗脱以及样品的记忆效应和/或进样口的污染可能也是间接法呈现较高过程空白的影响因素.但Coppola等[57 ] 的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素.Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... 另外,稳定碳同位素手段也可以用来初步鉴别CSRA是否受到明显外来碳的影响.假定外源碳和目标化合物的13 C值有明显的差别,通过比较稳定碳同位素比质谱仪测定的目标有机物单体δ13 C值和PCGC分离富集回收后的目标化合物的δ13 C值之间的差异,也可以评估是否存在有机溶剂残留或者是其他污染外源碳被引入[26 ,54 ] . ...
... 由于PCGC分离过程中目标化合物在高温条件下以气态的形式传输后进入捕集管,整个系统气密性的可持续性决定了目标化合物回收率的好坏,实际实验过程中PCGC的回收率一般在70%~80%[26 ,60 ] .在色谱分离过程中通常会发生同位素分馏,随着色谱信号的流出,其碳同位素组成是不断变化的[61 ,62 ] .因此,PCGC分离富集过程中目标化合物不完全回收往往也会导致一定的碳同位素分馏.Zencak等[54 ] 测定了香草醛标准不同截留时间馏分的δ13 C,结果显示其谱峰的前沿部分(-15.75‰)明显偏正于峰尾部分(-49.91‰),这与Eglinton等[26 ] 的结果一致.碳同位素呈现逆同位素效应(inverse isotope effect),即重的碳同位素先流出而轻的同位素后流出,这与Cl等同位素效应相反[63 ] .因此当目标化合物不完全分离或收集,会发生较为明显的碳同位素分馏.Zencak等[54 ] 同时指出,未经处理的香草醛δ13 C比值(-31.6‰)与经PCGC完整收集的香草醛δ13 C比值(-31.3‰)一致.但实际中,尤其是处理海洋沉积物等基质复杂的样品,为了获取高纯度的目标化合物,通常会缩短组分截留窗口的时间,即靠近目标化合物拖尾或前沿的部分截留,这可能导致目标化合物不完全回收而引起一定程度的碳同位素分馏.理论上,δ14 C的同位素分馏应当是δ13 C分馏的2倍,呈现和13 C同样的逆同位素效应.但是,放射性碳数据通常是报道经过δ13 C=-25‰校正后的Fm和Δ14 C值[64 ] .因此,即使发生碳同位素分馏,只会影响目标化合物的回收率及δ13 C数据的准确性,而不会影响最终报道的放射性碳同位素数据[64 ] . ...
... [26 ]的结果一致.碳同位素呈现逆同位素效应(inverse isotope effect),即重的碳同位素先流出而轻的同位素后流出,这与Cl等同位素效应相反[63 ] .因此当目标化合物不完全分离或收集,会发生较为明显的碳同位素分馏.Zencak等[54 ] 同时指出,未经处理的香草醛δ13 C比值(-31.6‰)与经PCGC完整收集的香草醛δ13 C比值(-31.3‰)一致.但实际中,尤其是处理海洋沉积物等基质复杂的样品,为了获取高纯度的目标化合物,通常会缩短组分截留窗口的时间,即靠近目标化合物拖尾或前沿的部分截留,这可能导致目标化合物不完全回收而引起一定程度的碳同位素分馏.理论上,δ14 C的同位素分馏应当是δ13 C分馏的2倍,呈现和13 C同样的逆同位素效应.但是,放射性碳数据通常是报道经过δ13 C=-25‰校正后的Fm和Δ14 C值[64 ] .因此,即使发生碳同位素分馏,只会影响目标化合物的回收率及δ13 C数据的准确性,而不会影响最终报道的放射性碳同位素数据[64 ] . ...
... 传统的PCGC配置了大孔径色谱柱(0.53 mm i.d.),提供了大负载容量的同时,牺牲了色谱的分离能力[43 ] .而为了弥补传统PCGC分离能力且减少或避免使用可能引入外源碳的其他湿化学萃取分离过程[38 ,54 ,57 ] ,有报道指出可以通过串联色谱及多维色谱技术提高传统PCGC对复杂基质的分离能力,同时也减少了繁琐的化学分离前处理过程中潜在外源碳的引入[42 ] .Ball等[42 ] 将二维PCGC应用于CSRA样品的分离.与传统PCGC相比,其增加了一个中心切割微流体切换装置(Deans pneumatic microfluidic switching device)以及串联2个不同固定相的色谱柱(Rtx-5 MS, 0.50 μm df和DB-17 MS, 0.25 μm df).尽管增加了微流体切换装置及色谱柱数量,但其保留时间漂移程度很小(标准偏差最大为0.016 min),这与传统PCGC的性能基本一致[26 ] .而多次进样发现其FID峰面积的偏差为1.1%~1.2%[42 ] ,这可能是与PTV进样模式所引起的扰动有关.传统PCGC柱流失对过程空白的贡献是(0.6±0.3) μg C/min,但如果无溶剂空运行PCGC,则柱流失仅贡献了0.1 μg C/min[38 ] .而在二维PCGC中,2°色谱柱仅仅从1°柱中接收有限量的样品,而并没有溶剂进入2°柱,因此柱流失并不是二维PCGC技术外源碳的重要来源.另外,在二维PCGC中使用了较薄的(0.25 μm, df)和低柱流失的固定相,可以进一步降低柱流失的贡献.Ball等[42 ] 通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定. ...
... 自从Eglinton等[26 ] 首次将单体分子放射性碳同位素分析技术应用于生物标志物14 C研究以来,该技术发展迅速,越来越多地应用于海洋科学、环境科学、生物地球化学和古气候学等领域,尤其是研究碳源和碳循环有关的问题.并且,随着高灵敏度AMS的快速发展,已经实现了超微样品14 C浓度的测试,甚至于1 μg C的测试[40 ] .但是,目前CSRA技术发展的最大限制是如何从复杂的自然环境样品中分离和收集纯的、大量的单体分子化合物,同时减少CSRA过程中引入的外源碳的污染[37 ,53 ] .近些年来,不断改进的PCGC和HPLC技术极大地扩展了生物标志物的应用,同时也提高了目标化合物分离的纯度和回收效率.目前,利用PCGC和Prep-HPLC等制备色谱进行分离和富集生物标志物单体时,对过程空白贡献的外源碳是1~2 μg C,因此,为了确保干扰物质低于10%,至少需要富集10~20 μg C以上的生物标志物单体进行其14 C测试.但据已有研究指出,CSRA技术过程空白具有高度的时空不确定性,所以将来的研究将需要更加关注“零外源碳”的分离纯化过程和严格的质量控制. ...
Variability in radiocarbon ages of individual organic compounds from marine sediments
2
1997
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... 自然环境研究中常用生物标志物的含量一般很低(一般小于1 μg/g干重,甚至低于几十个ng/g干重),所以需要通过分离和富集从自然环境样品中获取足够量(>100 μg C)、高纯度的生物标志物以满足AMS的测试要求.为了消除天然环境样品中复杂的基质干扰,首先会利用湿化学法进行目标组分初步的有机溶剂萃取和分离提纯.有机溶剂萃取技术包括超声萃取[44 ,45 ] 、索氏提取[27 ,46 ] 、加速溶剂萃取[47 ,48 ] 以及微波萃取[14 ,17 ] 等技术.在提纯生物标志物过程中,还需要配合一系列的化学分离技术和固相萃取技术等手段.根据化合物的不同化学性质,如中性组分(包括烷烃、醇类和烯酮等)和酸性组分(包括脂肪酸等),可以通过碱水解(0.5 mol/L的KOH的甲醇溶液)将二者分离[49 ] .固相萃取分离技术则是根据不同目标化合物的极性或结构差异实现进一步分离和提纯的化学法.例如,硅胶层析通过改变淋洗液极性梯度,可将不同极性的化合物分离[49 ] ;硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
A new look at old carbon in active margin sediments
1
2009
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
Widespread release of old carbon across the Siberian Arctic echoed by its large rivers
0
2011
14 C and 13 C characteristics of higher plant biomarkers in Washington margin surface sediments
10
2013
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... [30 ,42 ,43 ].本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... 自然环境研究中常用生物标志物的含量一般很低(一般小于1 μg/g干重,甚至低于几十个ng/g干重),所以需要通过分离和富集从自然环境样品中获取足够量(>100 μg C)、高纯度的生物标志物以满足AMS的测试要求.为了消除天然环境样品中复杂的基质干扰,首先会利用湿化学法进行目标组分初步的有机溶剂萃取和分离提纯.有机溶剂萃取技术包括超声萃取[44 ,45 ] 、索氏提取[27 ,46 ] 、加速溶剂萃取[47 ,48 ] 以及微波萃取[14 ,17 ] 等技术.在提纯生物标志物过程中,还需要配合一系列的化学分离技术和固相萃取技术等手段.根据化合物的不同化学性质,如中性组分(包括烷烃、醇类和烯酮等)和酸性组分(包括脂肪酸等),可以通过碱水解(0.5 mol/L的KOH的甲醇溶液)将二者分离[49 ] .固相萃取分离技术则是根据不同目标化合物的极性或结构差异实现进一步分离和提纯的化学法.例如,硅胶层析通过改变淋洗液极性梯度,可将不同极性的化合物分离[49 ] ;硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
... [30 ,49 ].然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
... 传统的PCGC配置了大孔径色谱柱(0.53 mm i.d.),提供了大负载容量的同时,牺牲了色谱的分离能力[43 ] .而为了弥补传统PCGC分离能力且减少或避免使用可能引入外源碳的其他湿化学萃取分离过程[38 ,54 ,57 ] ,有报道指出可以通过串联色谱及多维色谱技术提高传统PCGC对复杂基质的分离能力,同时也减少了繁琐的化学分离前处理过程中潜在外源碳的引入[42 ] .Ball等[42 ] 将二维PCGC应用于CSRA样品的分离.与传统PCGC相比,其增加了一个中心切割微流体切换装置(Deans pneumatic microfluidic switching device)以及串联2个不同固定相的色谱柱(Rtx-5 MS, 0.50 μm df和DB-17 MS, 0.25 μm df).尽管增加了微流体切换装置及色谱柱数量,但其保留时间漂移程度很小(标准偏差最大为0.016 min),这与传统PCGC的性能基本一致[26 ] .而多次进样发现其FID峰面积的偏差为1.1%~1.2%[42 ] ,这可能是与PTV进样模式所引起的扰动有关.传统PCGC柱流失对过程空白的贡献是(0.6±0.3) μg C/min,但如果无溶剂空运行PCGC,则柱流失仅贡献了0.1 μg C/min[38 ] .而在二维PCGC中,2°色谱柱仅仅从1°柱中接收有限量的样品,而并没有溶剂进入2°柱,因此柱流失并不是二维PCGC技术外源碳的重要来源.另外,在二维PCGC中使用了较薄的(0.25 μm, df)和低柱流失的固定相,可以进一步降低柱流失的贡献.Ball等[42 ] 通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定. ...
... Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段.Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致.可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标.Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右.但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
... Prep-HPLC技术被广泛应用于分离纯化环境样品中一类重要的陆源生物标志物——木质素.Ingalls等[39 ] 尝试利用双色谱柱制备液相色谱分离提纯了Pacific Northwest的叶片中的6种木质素酚类化合物.首先,利用反相-HPLC柱(Zorbax Eclipse XDB-C18 column,4.6 mm×150 mm; 5 μm)分离获得半纯的木质素酚类化合物,然后使用正相-HPLC柱(LiChrospher Diol column, 4.6 mm×150 mm, 5 μm)分离和收集纯的木质素酚类化合物单体.因为反相-HPLC不能完全分离所有的木质素酚类化合物,而利用正相-HPLC可以进一步分离在反相-HPLC中会共同溶出的木质素酚类化合物以及与目标木质素酚类化合物具有相似色谱行为的其他基质,从而获得纯度99%以上的目标化合物.相对于双色谱柱-HPLC技术,Hou等[14 ] 利用一个单柱(XDB-C18 )-HPLC技术成功分离了湖泊沉积物中的木质素,该方法在进行HPLC分离之前利用一个C18 -SPE纯化柱去除湖泊沉积物中的中性组分.Feng等[30 ] 通过固相萃取柱结合双色谱柱制备液相色谱分离提纯了华盛顿边缘海沉积物中木质素氧化产物.海洋沉积物尤其是边缘海区域,沉积有机质来源广泛、组成复杂、基质高,因此,相应的木质素酚类化合物分离提纯步骤较为繁琐.首先,通过ENVI-18固相萃取柱将中性化合物等不纯物质去除,然后通过LC-NH2 固相萃取柱将木质素酚类组分为醛/酮类组分和酸性组分.化学纯化后的木质素酚类组分通过Prep-HPLC分离收集用于之后的单体14 C同位素分析.使用Phenomenex Polar-RP柱(4.6 mm×250 mm, 4 μm)进行初步的分离收集,然后使用ZORBAX Eclipse XDB-C18 (4.6 mm×250 mm, 5 μm)进一步的分离纯化.尽管Feng等[30 ] 增加了分离纯化的步骤,但HPLC分离后,目标化合物的回收率(60%~80%)不低于已有报道的回收效率(约为50%)[39 ] ,加之木质素酚类化合物具有高的挥发性,因此导致其在柔和氮气吹干溶剂过程发生较大损失[39 ] . ...
... [30 ]增加了分离纯化的步骤,但HPLC分离后,目标化合物的回收率(60%~80%)不低于已有报道的回收效率(约为50%)[39 ] ,加之木质素酚类化合物具有高的挥发性,因此导致其在柔和氮气吹干溶剂过程发生较大损失[39 ] . ...
... Ingalls等[39 ] 评估了Prep-HPLC分离提纯木质素酚类化合物过程中引入的外源碳约有2.2 μg C((1.8±0.9) μg C化石源碳和0.35 μg C现代源碳),接近于其他HPLC分离过程产生的外源碳的贡献[14 ,30 ] .其中化石源碳的贡献大于80%,主要来自在微波消解Teflon管和/或聚碳酸酯离心管残留中[39 ] .另外,Feng等[30 ] 利用HPLC和PCGC技术分离的同一样品的Δ14 C结果显示,HPLC分离的香草基酚类化合物的Δ14 C值相比PCGC方法偏负21‰~29‰,二者的差异低于AMS的测试误差.但相较于PCGC方法,HPLC方法避免了大量使用衍生化试剂,且具有进样量大、效率高和低空白的优势. ...
... [30 ]利用HPLC和PCGC技术分离的同一样品的Δ14 C结果显示,HPLC分离的香草基酚类化合物的Δ14 C值相比PCGC方法偏负21‰~29‰,二者的差异低于AMS的测试误差.但相较于PCGC方法,HPLC方法避免了大量使用衍生化试剂,且具有进样量大、效率高和低空白的优势. ...
Differential mobilization of terrestrial carbon pools in Eurasian Arctic river basins
1
2013
... 另外,生物标志物单体14 C同位素也是一类常见的来源示踪指标,被广泛用于大气颗粒物、土壤、河流、湖泊、海洋等生态环境系统中有机碳的来源解析研究中[15 ~20 ] .由于高的初级生产力以及高的陆源输入,边缘海不仅是重要的海洋碳汇,也是一个重要的陆源碳汇.有机碳储库(carbon pools)(颗粒有机碳汇和溶解有机碳汇)中不同来源有机质的比例不仅决定了其组成性质和埋藏效率,而且还在不同尺度上影响全球碳循环.通过生物标志物单体14 C同位素技术可以将自然环境系统中有机质来源从“年龄”的角度基本可划分为三大类,包括年轻的海源有机质和陆源维管植物残留物(Δ14 C=0±50‰,近现代: <100年)、陈化的土壤有机质(千年尺度)及古老的沉积岩风化有机质(Δ14 C=-1 000‰,万年尺度)[21 ,22 ] .以上3种有机质中只有海洋生源和植被源有机碳汇能够影响短时间尺度大气CO2 的浓度,而陈化土壤源和化石源古老有机质常常会在陆地湖泊、土壤或沉积岩等中间有机碳储库中埋藏成百上千年甚至百万年,随时间的推移,通过河流或者风尘等传输过程进入海洋沉积环境中,所以这些陆源有机质的埋藏在短时间尺度上对近现代大气CO2 的埋藏贡献几乎为零.因此,通过14 C同位素特征将现代有机碳和其他年龄的有机碳区分开,有助于正确评估现代海洋“碳汇”的功能,并从“年龄”的新视角认识有机碳的迁移、转化和埋藏特征.大部分自然样品中有机碳的来源复杂,这一异质性导致总有机质的表观14 C同位素组成代表的是一个混合的信号.尤其是陈化土壤有机质的Δ14 C端元值的高度时空可变性[23 ,24 ] ,很难通过在自然界中找到对应的典型样品来确定它,导致总有机质14 C同位素信号无法区分和剥离较老的土壤源有机质和古老的化石源有机质的信号.由于有机物的14 C同位素特征不依赖于生物合成方式和化合物种类,只受合成时利用的碳源的14 C同位素水平和“中间储库”停留时间控制.生物标志物由特定生物合成,性质稳定,在分子水平上指示其特定来源[25 ] ,生物标志物14 C同位素组成被广泛应用于评价生物圈中有机质的来源及组成变化[16 ,26 ~31 ] .因此,通过来源具有专一性(Source Specific Characteristics)不同生物标志物单体14 C信号可以准确获取相应来源有机质的14 C同位素特征端元值,以达到示踪这类有机物“源—汇”过程变化. ...
Direct application of compound-specific radiocarbon analysis of leaf waxes to establish lacustrine sediment chronology
1
2008
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
Compound specific radiocarbon and δ13 C measurements of fatty acids in a continental aerosol sample
1
2001
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
Modern and fossil contributions to polycyclic aromatic hydrocarbons in PM2.5 from north Birmingham, Alabama in the southeastern U.S
1
2012
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
Determination of microbial carbon sources and cycling during remediation of petroleum hydrocarbon impacted soil using natural abundance 14 C analysis of PLFA
1
2010
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
A gas ion source for radiocarbon measurements at 200 kV
2
2007
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... 天然样品的CSRA分析中,PCGC是最早发展起来的用于同位素分析的高纯度单体制备色谱技术.PCGC具有高的分离能力,克服了毛细管气相色谱的有限容量(<500 ng/compound,compound为单个化合物)的技术限制.整个装置由2部分组成:气相色谱和Gerstel八通阀回收装置,共6个回收窗口(图1 ).气相色谱端耦合了CIS系统程序升温的PTV进样口、大口径色谱柱(0.53 mm(内径,i, d),0.5 μm(固定相膜厚,film thickness))、FID检测器、十字分流阀.溶剂排空方式的PTV进样口和大孔径色谱柱的使用可实现大体积(≥5 μL/injection)、大容量(约2 μg/compound)进样,通过30~50次的重复进样可以收集足够量的目标化合物(60~100 μg/compound)满足现代高灵敏度AMS[50 ,51 ] 的14 C同位素测定需要.经过高效气相色谱柱分离的目标有机化合物单体通过十字分流阀,约1%进入FID检测器,其余99%通过毛细管传输线(transfer line,0.32 mm(内径,i.d.))进入Gerstel八通阀回收装置;低的保留时间漂移程度(标准偏差最大为0.03 min)保证目标化合物多次收集的准确性和可靠性[26 ] .Eglinton等[26 ] 首次应用PCGC分离富集了海洋沉积物中常见的类脂生物标志物单体,实现了自然环境浓度水平的单体14 C测定.然而CSRA技术仍然遇到很多的挑战和限制,导致其还没有真正实现全自动化,甚至相关的质量控制标准也未建立.在自然环境研究中,CSRA技术的应用所面临的挑战和瓶颈主要是环境样品中常见的几类生物标志物含量都较低,其有机化合物单体的纯化制备过程复杂繁琐且技术要求高.随着高灵敏度加速器质谱的发展,14 C同位素测定所需的样品量越来越小,甚至能够测试最低至几个微克碳的样品[36 ,52 ] .因此,过程空白或外源碳(Cex )对样品尤其是小样品最终14 C结果产生的影响就愈加明显[37 ,40 ,53 ] . ...
Ultra-microscale (5-25 μg C) analysis of individual lipids by 14 C AMS: Assessment and correction for sample processing blanks
5
2007
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... [37 ,40 ].因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... 天然样品的CSRA分析中,PCGC是最早发展起来的用于同位素分析的高纯度单体制备色谱技术.PCGC具有高的分离能力,克服了毛细管气相色谱的有限容量(<500 ng/compound,compound为单个化合物)的技术限制.整个装置由2部分组成:气相色谱和Gerstel八通阀回收装置,共6个回收窗口(图1 ).气相色谱端耦合了CIS系统程序升温的PTV进样口、大口径色谱柱(0.53 mm(内径,i, d),0.5 μm(固定相膜厚,film thickness))、FID检测器、十字分流阀.溶剂排空方式的PTV进样口和大孔径色谱柱的使用可实现大体积(≥5 μL/injection)、大容量(约2 μg/compound)进样,通过30~50次的重复进样可以收集足够量的目标化合物(60~100 μg/compound)满足现代高灵敏度AMS[50 ,51 ] 的14 C同位素测定需要.经过高效气相色谱柱分离的目标有机化合物单体通过十字分流阀,约1%进入FID检测器,其余99%通过毛细管传输线(transfer line,0.32 mm(内径,i.d.))进入Gerstel八通阀回收装置;低的保留时间漂移程度(标准偏差最大为0.03 min)保证目标化合物多次收集的准确性和可靠性[26 ] .Eglinton等[26 ] 首次应用PCGC分离富集了海洋沉积物中常见的类脂生物标志物单体,实现了自然环境浓度水平的单体14 C测定.然而CSRA技术仍然遇到很多的挑战和限制,导致其还没有真正实现全自动化,甚至相关的质量控制标准也未建立.在自然环境研究中,CSRA技术的应用所面临的挑战和瓶颈主要是环境样品中常见的几类生物标志物含量都较低,其有机化合物单体的纯化制备过程复杂繁琐且技术要求高.随着高灵敏度加速器质谱的发展,14 C同位素测定所需的样品量越来越小,甚至能够测试最低至几个微克碳的样品[36 ,52 ] .因此,过程空白或外源碳(Cex )对样品尤其是小样品最终14 C结果产生的影响就愈加明显[37 ,40 ,53 ] . ...
... 随着CSRA技术在自然环境样品中的应用,样品量逐渐向超微样品(<25 μg C)发展,此时过程空白也就成为了CSRA样品14 C测试精确度和准确度的实际限制因素[37 ,56 ] .自然环境中的样品,化学萃取和PCGC分离富集过程中引入的外源碳是过程空白的主要贡献,远远高于在真空线转换为CO2 和/或石墨化过程中外源碳的量;并且当样品量变小时(<100 μg C),过程空白的评价显得更为重要[40 ] .在PCGC分离过程中,色谱柱流失是外源碳的主要来源之一.Eglinton等[26 ] 发现,当PCGC使用高容量色谱柱(>1 μm固定相膜厚)时,尤其是高沸点的有机化合物单体组分中,GC-FID能够明显从PCGC-U型捕集管中检测出聚甲基硅酮的降解产物;而对于使用较薄的固定相时(≤0.5 μm),色谱柱流失的影响则明显减少. ...
... 自从Eglinton等[26 ] 首次将单体分子放射性碳同位素分析技术应用于生物标志物14 C研究以来,该技术发展迅速,越来越多地应用于海洋科学、环境科学、生物地球化学和古气候学等领域,尤其是研究碳源和碳循环有关的问题.并且,随着高灵敏度AMS的快速发展,已经实现了超微样品14 C浓度的测试,甚至于1 μg C的测试[40 ] .但是,目前CSRA技术发展的最大限制是如何从复杂的自然环境样品中分离和收集纯的、大量的单体分子化合物,同时减少CSRA过程中引入的外源碳的污染[37 ,53 ] .近些年来,不断改进的PCGC和HPLC技术极大地扩展了生物标志物的应用,同时也提高了目标化合物分离的纯度和回收效率.目前,利用PCGC和Prep-HPLC等制备色谱进行分离和富集生物标志物单体时,对过程空白贡献的外源碳是1~2 μg C,因此,为了确保干扰物质低于10%,至少需要富集10~20 μg C以上的生物标志物单体进行其14 C测试.但据已有研究指出,CSRA技术过程空白具有高度的时空不确定性,所以将来的研究将需要更加关注“零外源碳”的分离纯化过程和严格的质量控制. ...
Druffel E R M. Quantification of extraneous carbon during compound specific radiocarbon analysis of black carbon
8
2009
... 定量估算PCGC分离过程所引入外源碳的方法分别为直接法(directly method)和间接法(indirectly method)[38 ,57 ] .直接法是假定在PCGC分离过程中的外源碳主要是来自柱流失(包括过去样品的残留和/或GC色谱柱固定相的脱落),且柱流失不随时间和温度的变化而改变.在无溶剂条件下运行PCGC,馏分收集器按照目标化合物的截留时间收集相同条件下的仪器本底空白.Ziolkowski等[38 ] 通过直接法评估了PCGC分离过程中空白的质量随馏分收集器的收集窗口时间增加而增加,平均产生外源碳为(0.1±0.05) μg C /min(Fm=0.125±0.034).而间接方法则是通过加入已知14 C同位素水平的参考标准——“现代源(modern)”有机化合物标准(Fm=1)和“化石源(fossil)”有机化合物标准(Fm=0),分别评估PCGC分离过程中引入的化石源空白碳和现代空白碳的质量.二者质量加和即为引入的外源碳空白总质量,二者Fm的质量加权平均值即为外源碳空白的同位素特征值[38 ] .Ziolkowski等[38 ] 的研究结果显示通过间接法评估得到的PCGC分离过程中引入的外源碳中化石源空白碳和现代源空白碳的贡献分别是Cpcgc-fossil =(0.4±0.2) μg C/min和Cpcgc-modern =(0.2±0.1) μg C/min.因此,应用间接方法估算的外源碳的贡献是Cpcgc =(0.6±0.3) μg C/min(Fm=0.3±0.1).2种方法估算的PCGC分离过程中引入外源碳的贡献相差较大(0.5 μg C),这可能是在有溶剂条件下,来自色谱柱的外源碳更容易被洗脱以及样品的记忆效应和/或进样口的污染可能也是间接法呈现较高过程空白的影响因素.但Coppola等[57 ] 的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素.Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... [38 ]通过直接法评估了PCGC分离过程中空白的质量随馏分收集器的收集窗口时间增加而增加,平均产生外源碳为(0.1±0.05) μg C /min(Fm=0.125±0.034).而间接方法则是通过加入已知14 C同位素水平的参考标准——“现代源(modern)”有机化合物标准(Fm=1)和“化石源(fossil)”有机化合物标准(Fm=0),分别评估PCGC分离过程中引入的化石源空白碳和现代空白碳的质量.二者质量加和即为引入的外源碳空白总质量,二者Fm的质量加权平均值即为外源碳空白的同位素特征值[38 ] .Ziolkowski等[38 ] 的研究结果显示通过间接法评估得到的PCGC分离过程中引入的外源碳中化石源空白碳和现代源空白碳的贡献分别是Cpcgc-fossil =(0.4±0.2) μg C/min和Cpcgc-modern =(0.2±0.1) μg C/min.因此,应用间接方法估算的外源碳的贡献是Cpcgc =(0.6±0.3) μg C/min(Fm=0.3±0.1).2种方法估算的PCGC分离过程中引入外源碳的贡献相差较大(0.5 μg C),这可能是在有溶剂条件下,来自色谱柱的外源碳更容易被洗脱以及样品的记忆效应和/或进样口的污染可能也是间接法呈现较高过程空白的影响因素.但Coppola等[57 ] 的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素.Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... [38 ].Ziolkowski等[38 ] 的研究结果显示通过间接法评估得到的PCGC分离过程中引入的外源碳中化石源空白碳和现代源空白碳的贡献分别是Cpcgc-fossil =(0.4±0.2) μg C/min和Cpcgc-modern =(0.2±0.1) μg C/min.因此,应用间接方法估算的外源碳的贡献是Cpcgc =(0.6±0.3) μg C/min(Fm=0.3±0.1).2种方法估算的PCGC分离过程中引入外源碳的贡献相差较大(0.5 μg C),这可能是在有溶剂条件下,来自色谱柱的外源碳更容易被洗脱以及样品的记忆效应和/或进样口的污染可能也是间接法呈现较高过程空白的影响因素.但Coppola等[57 ] 的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素.Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... [38 ]的研究结果显示通过间接法评估得到的PCGC分离过程中引入的外源碳中化石源空白碳和现代源空白碳的贡献分别是Cpcgc-fossil =(0.4±0.2) μg C/min和Cpcgc-modern =(0.2±0.1) μg C/min.因此,应用间接方法估算的外源碳的贡献是Cpcgc =(0.6±0.3) μg C/min(Fm=0.3±0.1).2种方法估算的PCGC分离过程中引入外源碳的贡献相差较大(0.5 μg C),这可能是在有溶剂条件下,来自色谱柱的外源碳更容易被洗脱以及样品的记忆效应和/或进样口的污染可能也是间接法呈现较高过程空白的影响因素.但Coppola等[57 ] 的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素.Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... [38 ]的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... [38 ]同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... 传统的PCGC配置了大孔径色谱柱(0.53 mm i.d.),提供了大负载容量的同时,牺牲了色谱的分离能力[43 ] .而为了弥补传统PCGC分离能力且减少或避免使用可能引入外源碳的其他湿化学萃取分离过程[38 ,54 ,57 ] ,有报道指出可以通过串联色谱及多维色谱技术提高传统PCGC对复杂基质的分离能力,同时也减少了繁琐的化学分离前处理过程中潜在外源碳的引入[42 ] .Ball等[42 ] 将二维PCGC应用于CSRA样品的分离.与传统PCGC相比,其增加了一个中心切割微流体切换装置(Deans pneumatic microfluidic switching device)以及串联2个不同固定相的色谱柱(Rtx-5 MS, 0.50 μm df和DB-17 MS, 0.25 μm df).尽管增加了微流体切换装置及色谱柱数量,但其保留时间漂移程度很小(标准偏差最大为0.016 min),这与传统PCGC的性能基本一致[26 ] .而多次进样发现其FID峰面积的偏差为1.1%~1.2%[42 ] ,这可能是与PTV进样模式所引起的扰动有关.传统PCGC柱流失对过程空白的贡献是(0.6±0.3) μg C/min,但如果无溶剂空运行PCGC,则柱流失仅贡献了0.1 μg C/min[38 ] .而在二维PCGC中,2°色谱柱仅仅从1°柱中接收有限量的样品,而并没有溶剂进入2°柱,因此柱流失并不是二维PCGC技术外源碳的重要来源.另外,在二维PCGC中使用了较薄的(0.25 μm, df)和低柱流失的固定相,可以进一步降低柱流失的贡献.Ball等[42 ] 通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定. ...
... [38 ].而在二维PCGC中,2°色谱柱仅仅从1°柱中接收有限量的样品,而并没有溶剂进入2°柱,因此柱流失并不是二维PCGC技术外源碳的重要来源.另外,在二维PCGC中使用了较薄的(0.25 μm, df)和低柱流失的固定相,可以进一步降低柱流失的贡献.Ball等[42 ] 通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定. ...
HPLC purification of higher plant-dervied lignin phenols for compound specific radiocarbon analysis
6
2010
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... Prep-HPLC技术被广泛应用于分离纯化环境样品中一类重要的陆源生物标志物——木质素.Ingalls等[39 ] 尝试利用双色谱柱制备液相色谱分离提纯了Pacific Northwest的叶片中的6种木质素酚类化合物.首先,利用反相-HPLC柱(Zorbax Eclipse XDB-C18 column,4.6 mm×150 mm; 5 μm)分离获得半纯的木质素酚类化合物,然后使用正相-HPLC柱(LiChrospher Diol column, 4.6 mm×150 mm, 5 μm)分离和收集纯的木质素酚类化合物单体.因为反相-HPLC不能完全分离所有的木质素酚类化合物,而利用正相-HPLC可以进一步分离在反相-HPLC中会共同溶出的木质素酚类化合物以及与目标木质素酚类化合物具有相似色谱行为的其他基质,从而获得纯度99%以上的目标化合物.相对于双色谱柱-HPLC技术,Hou等[14 ] 利用一个单柱(XDB-C18 )-HPLC技术成功分离了湖泊沉积物中的木质素,该方法在进行HPLC分离之前利用一个C18 -SPE纯化柱去除湖泊沉积物中的中性组分.Feng等[30 ] 通过固相萃取柱结合双色谱柱制备液相色谱分离提纯了华盛顿边缘海沉积物中木质素氧化产物.海洋沉积物尤其是边缘海区域,沉积有机质来源广泛、组成复杂、基质高,因此,相应的木质素酚类化合物分离提纯步骤较为繁琐.首先,通过ENVI-18固相萃取柱将中性化合物等不纯物质去除,然后通过LC-NH2 固相萃取柱将木质素酚类组分为醛/酮类组分和酸性组分.化学纯化后的木质素酚类组分通过Prep-HPLC分离收集用于之后的单体14 C同位素分析.使用Phenomenex Polar-RP柱(4.6 mm×250 mm, 4 μm)进行初步的分离收集,然后使用ZORBAX Eclipse XDB-C18 (4.6 mm×250 mm, 5 μm)进一步的分离纯化.尽管Feng等[30 ] 增加了分离纯化的步骤,但HPLC分离后,目标化合物的回收率(60%~80%)不低于已有报道的回收效率(约为50%)[39 ] ,加之木质素酚类化合物具有高的挥发性,因此导致其在柔和氮气吹干溶剂过程发生较大损失[39 ] . ...
... [39 ],加之木质素酚类化合物具有高的挥发性,因此导致其在柔和氮气吹干溶剂过程发生较大损失[39 ] . ...
... [39 ]. ...
... Ingalls等[39 ] 评估了Prep-HPLC分离提纯木质素酚类化合物过程中引入的外源碳约有2.2 μg C((1.8±0.9) μg C化石源碳和0.35 μg C现代源碳),接近于其他HPLC分离过程产生的外源碳的贡献[14 ,30 ] .其中化石源碳的贡献大于80%,主要来自在微波消解Teflon管和/或聚碳酸酯离心管残留中[39 ] .另外,Feng等[30 ] 利用HPLC和PCGC技术分离的同一样品的Δ14 C结果显示,HPLC分离的香草基酚类化合物的Δ14 C值相比PCGC方法偏负21‰~29‰,二者的差异低于AMS的测试误差.但相较于PCGC方法,HPLC方法避免了大量使用衍生化试剂,且具有进样量大、效率高和低空白的优势. ...
... [39 ].另外,Feng等[30 ] 利用HPLC和PCGC技术分离的同一样品的Δ14 C结果显示,HPLC分离的香草基酚类化合物的Δ14 C值相比PCGC方法偏负21‰~29‰,二者的差异低于AMS的测试误差.但相较于PCGC方法,HPLC方法避免了大量使用衍生化试剂,且具有进样量大、效率高和低空白的优势. ...
Glacial-interglacial variability in Atlantic meridional overturning circulation and thermocline adjustments in the tropical North Atlantic
4
2010
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... 天然样品的CSRA分析中,PCGC是最早发展起来的用于同位素分析的高纯度单体制备色谱技术.PCGC具有高的分离能力,克服了毛细管气相色谱的有限容量(<500 ng/compound,compound为单个化合物)的技术限制.整个装置由2部分组成:气相色谱和Gerstel八通阀回收装置,共6个回收窗口(图1 ).气相色谱端耦合了CIS系统程序升温的PTV进样口、大口径色谱柱(0.53 mm(内径,i, d),0.5 μm(固定相膜厚,film thickness))、FID检测器、十字分流阀.溶剂排空方式的PTV进样口和大孔径色谱柱的使用可实现大体积(≥5 μL/injection)、大容量(约2 μg/compound)进样,通过30~50次的重复进样可以收集足够量的目标化合物(60~100 μg/compound)满足现代高灵敏度AMS[50 ,51 ] 的14 C同位素测定需要.经过高效气相色谱柱分离的目标有机化合物单体通过十字分流阀,约1%进入FID检测器,其余99%通过毛细管传输线(transfer line,0.32 mm(内径,i.d.))进入Gerstel八通阀回收装置;低的保留时间漂移程度(标准偏差最大为0.03 min)保证目标化合物多次收集的准确性和可靠性[26 ] .Eglinton等[26 ] 首次应用PCGC分离富集了海洋沉积物中常见的类脂生物标志物单体,实现了自然环境浓度水平的单体14 C测定.然而CSRA技术仍然遇到很多的挑战和限制,导致其还没有真正实现全自动化,甚至相关的质量控制标准也未建立.在自然环境研究中,CSRA技术的应用所面临的挑战和瓶颈主要是环境样品中常见的几类生物标志物含量都较低,其有机化合物单体的纯化制备过程复杂繁琐且技术要求高.随着高灵敏度加速器质谱的发展,14 C同位素测定所需的样品量越来越小,甚至能够测试最低至几个微克碳的样品[36 ,52 ] .因此,过程空白或外源碳(Cex )对样品尤其是小样品最终14 C结果产生的影响就愈加明显[37 ,40 ,53 ] . ...
... 随着CSRA技术在自然环境样品中的应用,样品量逐渐向超微样品(<25 μg C)发展,此时过程空白也就成为了CSRA样品14 C测试精确度和准确度的实际限制因素[37 ,56 ] .自然环境中的样品,化学萃取和PCGC分离富集过程中引入的外源碳是过程空白的主要贡献,远远高于在真空线转换为CO2 和/或石墨化过程中外源碳的量;并且当样品量变小时(<100 μg C),过程空白的评价显得更为重要[40 ] .在PCGC分离过程中,色谱柱流失是外源碳的主要来源之一.Eglinton等[26 ] 发现,当PCGC使用高容量色谱柱(>1 μm固定相膜厚)时,尤其是高沸点的有机化合物单体组分中,GC-FID能够明显从PCGC-U型捕集管中检测出聚甲基硅酮的降解产物;而对于使用较薄的固定相时(≤0.5 μm),色谱柱流失的影响则明显减少. ...
... 自从Eglinton等[26 ] 首次将单体分子放射性碳同位素分析技术应用于生物标志物14 C研究以来,该技术发展迅速,越来越多地应用于海洋科学、环境科学、生物地球化学和古气候学等领域,尤其是研究碳源和碳循环有关的问题.并且,随着高灵敏度AMS的快速发展,已经实现了超微样品14 C浓度的测试,甚至于1 μg C的测试[40 ] .但是,目前CSRA技术发展的最大限制是如何从复杂的自然环境样品中分离和收集纯的、大量的单体分子化合物,同时减少CSRA过程中引入的外源碳的污染[37 ,53 ] .近些年来,不断改进的PCGC和HPLC技术极大地扩展了生物标志物的应用,同时也提高了目标化合物分离的纯度和回收效率.目前,利用PCGC和Prep-HPLC等制备色谱进行分离和富集生物标志物单体时,对过程空白贡献的外源碳是1~2 μg C,因此,为了确保干扰物质低于10%,至少需要富集10~20 μg C以上的生物标志物单体进行其14 C测试.但据已有研究指出,CSRA技术过程空白具有高度的时空不确定性,所以将来的研究将需要更加关注“零外源碳”的分离纯化过程和严格的质量控制. ...
Optimization of a preparative capillary gas chromatography-mass spectrometry system for the isolation and harvesting of individual polycyclic aromatic hydrocarbons
1
2003
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
A two-dimensional, heart-cutting preparative gas chromatograph facilitates highly resolved single-compound isolations with utility towards compound-specific natural abundance radiocarbon (14 C) analyses
5
2012
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... 传统的PCGC配置了大孔径色谱柱(0.53 mm i.d.),提供了大负载容量的同时,牺牲了色谱的分离能力[43 ] .而为了弥补传统PCGC分离能力且减少或避免使用可能引入外源碳的其他湿化学萃取分离过程[38 ,54 ,57 ] ,有报道指出可以通过串联色谱及多维色谱技术提高传统PCGC对复杂基质的分离能力,同时也减少了繁琐的化学分离前处理过程中潜在外源碳的引入[42 ] .Ball等[42 ] 将二维PCGC应用于CSRA样品的分离.与传统PCGC相比,其增加了一个中心切割微流体切换装置(Deans pneumatic microfluidic switching device)以及串联2个不同固定相的色谱柱(Rtx-5 MS, 0.50 μm df和DB-17 MS, 0.25 μm df).尽管增加了微流体切换装置及色谱柱数量,但其保留时间漂移程度很小(标准偏差最大为0.016 min),这与传统PCGC的性能基本一致[26 ] .而多次进样发现其FID峰面积的偏差为1.1%~1.2%[42 ] ,这可能是与PTV进样模式所引起的扰动有关.传统PCGC柱流失对过程空白的贡献是(0.6±0.3) μg C/min,但如果无溶剂空运行PCGC,则柱流失仅贡献了0.1 μg C/min[38 ] .而在二维PCGC中,2°色谱柱仅仅从1°柱中接收有限量的样品,而并没有溶剂进入2°柱,因此柱流失并不是二维PCGC技术外源碳的重要来源.另外,在二维PCGC中使用了较薄的(0.25 μm, df)和低柱流失的固定相,可以进一步降低柱流失的贡献.Ball等[42 ] 通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定. ...
... [42 ]将二维PCGC应用于CSRA样品的分离.与传统PCGC相比,其增加了一个中心切割微流体切换装置(Deans pneumatic microfluidic switching device)以及串联2个不同固定相的色谱柱(Rtx-5 MS, 0.50 μm df和DB-17 MS, 0.25 μm df).尽管增加了微流体切换装置及色谱柱数量,但其保留时间漂移程度很小(标准偏差最大为0.016 min),这与传统PCGC的性能基本一致[26 ] .而多次进样发现其FID峰面积的偏差为1.1%~1.2%[42 ] ,这可能是与PTV进样模式所引起的扰动有关.传统PCGC柱流失对过程空白的贡献是(0.6±0.3) μg C/min,但如果无溶剂空运行PCGC,则柱流失仅贡献了0.1 μg C/min[38 ] .而在二维PCGC中,2°色谱柱仅仅从1°柱中接收有限量的样品,而并没有溶剂进入2°柱,因此柱流失并不是二维PCGC技术外源碳的重要来源.另外,在二维PCGC中使用了较薄的(0.25 μm, df)和低柱流失的固定相,可以进一步降低柱流失的贡献.Ball等[42 ] 通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定. ...
... [42 ],这可能是与PTV进样模式所引起的扰动有关.传统PCGC柱流失对过程空白的贡献是(0.6±0.3) μg C/min,但如果无溶剂空运行PCGC,则柱流失仅贡献了0.1 μg C/min[38 ] .而在二维PCGC中,2°色谱柱仅仅从1°柱中接收有限量的样品,而并没有溶剂进入2°柱,因此柱流失并不是二维PCGC技术外源碳的重要来源.另外,在二维PCGC中使用了较薄的(0.25 μm, df)和低柱流失的固定相,可以进一步降低柱流失的贡献.Ball等[42 ] 通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定. ...
... [42 ]通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定. ...
Improving the productivity of a multidimensional chromatographic preparative system by collecting pure chemicals after each of three chromatographic dimensions
2
2016
... Eglinton等[26 ] 首次应用制备气相色谱技术(Preparative Capillary Gas Chromatography, PCGC)发展了有机化合物单体放射性碳同位素分析技术(Compound-Specific Radiocarbon Analysis,CSRA).CSRA技术主要分3步:首先是将环境样品中的目标有机化合物进行化学预分离;然后通过制备色谱分离富集纯的有机化合物单体;最后利用离线的加速器质谱仪(Accelerator Mass Spectrometry, AMS)测定有机化合物单体14 C含量.通过该技术手段,Eglinton等[26 ] 成功地从海洋沉积物中分离并富集了高纯度(约99%)的正构烷烃及脂肪酸等生物标志物,开创了CSRA技术在自然环境14 C同位素水平研究中应用的先河.此后CSRA技术被广泛应用于海洋和湖泊沉积物[5 ,12 ,18 ,27 ,30 ,32 ] 、河流悬浮颗粒物[17 ,23 ,28 ] 、大气气溶胶[33 ,34 ] 和土壤[35 ] 等研究中.近些年,随着高灵敏度AMS的发展,低至3 μg C 的超微样品14 C的测试成为可能[36 ] ,极大地推动了CSRA技术在自然环境研究中的应用[37 ~39 ] .对于CSRA样品,尤其是超微样品(< 25 μg C),过程空白或外源碳的污染则是结果精确度和准确度的实际限制因素[37 ,40 ] .因为自然环境中目标生物标志物大多受基质干扰、难以分离,并且含量极低(μg/mL或更少),所以在进行单体14 C同位素测定前,需要通过复杂的化学萃取及色谱分离过程来富集高纯度的目标化合物,如何从基质组成复杂的样品中分离富集高纯度的目标化合物已经成为限制CSRA技术发展和应用的瓶颈[41 ] .除了传统化学分离提纯和PCGC分离技术,多维PCGC和制备液相色谱(Preparative High Performance Liquid Chromatography, Prep-HPLC)等技术也相继被发展和应用于自然环境样品的生物标志物分离富集提纯中[30 ,42 ,43 ] .本文旨在综合介绍自然环境研究中单体化合物放射性碳同位素分析技术中常见生物标志物单体分离纯化的技术方法以及发展现状. ...
... 传统的PCGC配置了大孔径色谱柱(0.53 mm i.d.),提供了大负载容量的同时,牺牲了色谱的分离能力[43 ] .而为了弥补传统PCGC分离能力且减少或避免使用可能引入外源碳的其他湿化学萃取分离过程[38 ,54 ,57 ] ,有报道指出可以通过串联色谱及多维色谱技术提高传统PCGC对复杂基质的分离能力,同时也减少了繁琐的化学分离前处理过程中潜在外源碳的引入[42 ] .Ball等[42 ] 将二维PCGC应用于CSRA样品的分离.与传统PCGC相比,其增加了一个中心切割微流体切换装置(Deans pneumatic microfluidic switching device)以及串联2个不同固定相的色谱柱(Rtx-5 MS, 0.50 μm df和DB-17 MS, 0.25 μm df).尽管增加了微流体切换装置及色谱柱数量,但其保留时间漂移程度很小(标准偏差最大为0.016 min),这与传统PCGC的性能基本一致[26 ] .而多次进样发现其FID峰面积的偏差为1.1%~1.2%[42 ] ,这可能是与PTV进样模式所引起的扰动有关.传统PCGC柱流失对过程空白的贡献是(0.6±0.3) μg C/min,但如果无溶剂空运行PCGC,则柱流失仅贡献了0.1 μg C/min[38 ] .而在二维PCGC中,2°色谱柱仅仅从1°柱中接收有限量的样品,而并没有溶剂进入2°柱,因此柱流失并不是二维PCGC技术外源碳的重要来源.另外,在二维PCGC中使用了较薄的(0.25 μm, df)和低柱流失的固定相,可以进一步降低柱流失的贡献.Ball等[42 ] 通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定. ...
Calibration of the alkenone paleotemperature index based on core-tops from the eastern South Atlantic and the global ocean (60°N~60°S)
1
1998
... 自然环境研究中常用生物标志物的含量一般很低(一般小于1 μg/g干重,甚至低于几十个ng/g干重),所以需要通过分离和富集从自然环境样品中获取足够量(>100 μg C)、高纯度的生物标志物以满足AMS的测试要求.为了消除天然环境样品中复杂的基质干扰,首先会利用湿化学法进行目标组分初步的有机溶剂萃取和分离提纯.有机溶剂萃取技术包括超声萃取[44 ,45 ] 、索氏提取[27 ,46 ] 、加速溶剂萃取[47 ,48 ] 以及微波萃取[14 ,17 ] 等技术.在提纯生物标志物过程中,还需要配合一系列的化学分离技术和固相萃取技术等手段.根据化合物的不同化学性质,如中性组分(包括烷烃、醇类和烯酮等)和酸性组分(包括脂肪酸等),可以通过碱水解(0.5 mol/L的KOH的甲醇溶液)将二者分离[49 ] .固相萃取分离技术则是根据不同目标化合物的极性或结构差异实现进一步分离和提纯的化学法.例如,硅胶层析通过改变淋洗液极性梯度,可将不同极性的化合物分离[49 ] ;硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
Terrestrial and marine biomarker estimates of organic matter sources and distributions in surface sediments from the East China Sea shelf
1
2011
... 自然环境研究中常用生物标志物的含量一般很低(一般小于1 μg/g干重,甚至低于几十个ng/g干重),所以需要通过分离和富集从自然环境样品中获取足够量(>100 μg C)、高纯度的生物标志物以满足AMS的测试要求.为了消除天然环境样品中复杂的基质干扰,首先会利用湿化学法进行目标组分初步的有机溶剂萃取和分离提纯.有机溶剂萃取技术包括超声萃取[44 ,45 ] 、索氏提取[27 ,46 ] 、加速溶剂萃取[47 ,48 ] 以及微波萃取[14 ,17 ] 等技术.在提纯生物标志物过程中,还需要配合一系列的化学分离技术和固相萃取技术等手段.根据化合物的不同化学性质,如中性组分(包括烷烃、醇类和烯酮等)和酸性组分(包括脂肪酸等),可以通过碱水解(0.5 mol/L的KOH的甲醇溶液)将二者分离[49 ] .固相萃取分离技术则是根据不同目标化合物的极性或结构差异实现进一步分离和提纯的化学法.例如,硅胶层析通过改变淋洗液极性梯度,可将不同极性的化合物分离[49 ] ;硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
Leaf wax lipids as paleovegetational and paleoenvironmental proxies for the Chinese Loess Plateau over the last 170 kyr
1
2006
... 自然环境研究中常用生物标志物的含量一般很低(一般小于1 μg/g干重,甚至低于几十个ng/g干重),所以需要通过分离和富集从自然环境样品中获取足够量(>100 μg C)、高纯度的生物标志物以满足AMS的测试要求.为了消除天然环境样品中复杂的基质干扰,首先会利用湿化学法进行目标组分初步的有机溶剂萃取和分离提纯.有机溶剂萃取技术包括超声萃取[44 ,45 ] 、索氏提取[27 ,46 ] 、加速溶剂萃取[47 ,48 ] 以及微波萃取[14 ,17 ] 等技术.在提纯生物标志物过程中,还需要配合一系列的化学分离技术和固相萃取技术等手段.根据化合物的不同化学性质,如中性组分(包括烷烃、醇类和烯酮等)和酸性组分(包括脂肪酸等),可以通过碱水解(0.5 mol/L的KOH的甲醇溶液)将二者分离[49 ] .固相萃取分离技术则是根据不同目标化合物的极性或结构差异实现进一步分离和提纯的化学法.例如,硅胶层析通过改变淋洗液极性梯度,可将不同极性的化合物分离[49 ] ;硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
Pressurized liquid extraction of selected molecular biomarkers in deep sea sediments used as proxies in paleoceanography
1
2003
... 自然环境研究中常用生物标志物的含量一般很低(一般小于1 μg/g干重,甚至低于几十个ng/g干重),所以需要通过分离和富集从自然环境样品中获取足够量(>100 μg C)、高纯度的生物标志物以满足AMS的测试要求.为了消除天然环境样品中复杂的基质干扰,首先会利用湿化学法进行目标组分初步的有机溶剂萃取和分离提纯.有机溶剂萃取技术包括超声萃取[44 ,45 ] 、索氏提取[27 ,46 ] 、加速溶剂萃取[47 ,48 ] 以及微波萃取[14 ,17 ] 等技术.在提纯生物标志物过程中,还需要配合一系列的化学分离技术和固相萃取技术等手段.根据化合物的不同化学性质,如中性组分(包括烷烃、醇类和烯酮等)和酸性组分(包括脂肪酸等),可以通过碱水解(0.5 mol/L的KOH的甲醇溶液)将二者分离[49 ] .固相萃取分离技术则是根据不同目标化合物的极性或结构差异实现进一步分离和提纯的化学法.例如,硅胶层析通过改变淋洗液极性梯度,可将不同极性的化合物分离[49 ] ;硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
Isolation and compound specific radiocarbon dating of terrigenous branched glycerol dialkyl glycerol tetraethers (brGDGTs)
4
2013
... 自然环境研究中常用生物标志物的含量一般很低(一般小于1 μg/g干重,甚至低于几十个ng/g干重),所以需要通过分离和富集从自然环境样品中获取足够量(>100 μg C)、高纯度的生物标志物以满足AMS的测试要求.为了消除天然环境样品中复杂的基质干扰,首先会利用湿化学法进行目标组分初步的有机溶剂萃取和分离提纯.有机溶剂萃取技术包括超声萃取[44 ,45 ] 、索氏提取[27 ,46 ] 、加速溶剂萃取[47 ,48 ] 以及微波萃取[14 ,17 ] 等技术.在提纯生物标志物过程中,还需要配合一系列的化学分离技术和固相萃取技术等手段.根据化合物的不同化学性质,如中性组分(包括烷烃、醇类和烯酮等)和酸性组分(包括脂肪酸等),可以通过碱水解(0.5 mol/L的KOH的甲醇溶液)将二者分离[49 ] .固相萃取分离技术则是根据不同目标化合物的极性或结构差异实现进一步分离和提纯的化学法.例如,硅胶层析通过改变淋洗液极性梯度,可将不同极性的化合物分离[49 ] ;硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
... Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段.Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致.可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标.Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右.但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
... Birhkolz等[48 ] 改进了已有的Prep-HPLC技术,成功分离富集出湖泊沉积物中br-GDGTs化合物单体.首先利用反相-HPLC(Agilent Eclipse Plus C18 column,4.6 mm×150 mm, 3.5 μm)将br-GDGTs和共流出组分分离;为了除去反相-HPLC柱分离过程中引入的共流出组分和进一步纯化,使用正相-HPLC柱(Alltech Prevail Cyano column, 2.1 mm×150 mm, 3 μm)消除可能来自流动相的外源碳(12 μg C/60 min).改进后的HPLC技术贡献的外源碳为 (0.6±0.2) μg C(其F14 C为0.5±0.2),与Hou等[14 ] 结果基本一致,其外源碳的贡献约为2 μg C(Fm值为0.30).基于br-GDGTs的BIT指标已被广泛用来估算海洋环境中土壤有机质的贡献比例[71 ~73 ] ,但有报道指出湖泊[74 ] 和海洋[75 ] 也会产生br-GDGTs.湖泊、海洋环境样品中br-GDGTs的14 C同位素特征可以为br-GDGTs土壤源指标的应用提供重要的信息.Birkholz等[48 ] 发现Lake Lusvatnet沉积物中的br-GDGTs呈现较老的14 C年龄.结合湖泊水体年轻的DOC特征[76 ] ,可明确判断该湖泊br-GDGTs来源于外源陈化土壤的输入. ...
... [48 ]发现Lake Lusvatnet沉积物中的br-GDGTs呈现较老的14 C年龄.结合湖泊水体年轻的DOC特征[76 ] ,可明确判断该湖泊br-GDGTs来源于外源陈化土壤的输入. ...
Radiocarbon dating of alkenones from marine sediments: I. Isolation protocol
4
2005
... 自然环境研究中常用生物标志物的含量一般很低(一般小于1 μg/g干重,甚至低于几十个ng/g干重),所以需要通过分离和富集从自然环境样品中获取足够量(>100 μg C)、高纯度的生物标志物以满足AMS的测试要求.为了消除天然环境样品中复杂的基质干扰,首先会利用湿化学法进行目标组分初步的有机溶剂萃取和分离提纯.有机溶剂萃取技术包括超声萃取[44 ,45 ] 、索氏提取[27 ,46 ] 、加速溶剂萃取[47 ,48 ] 以及微波萃取[14 ,17 ] 等技术.在提纯生物标志物过程中,还需要配合一系列的化学分离技术和固相萃取技术等手段.根据化合物的不同化学性质,如中性组分(包括烷烃、醇类和烯酮等)和酸性组分(包括脂肪酸等),可以通过碱水解(0.5 mol/L的KOH的甲醇溶液)将二者分离[49 ] .固相萃取分离技术则是根据不同目标化合物的极性或结构差异实现进一步分离和提纯的化学法.例如,硅胶层析通过改变淋洗液极性梯度,可将不同极性的化合物分离[49 ] ;硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
... [49 ];硝酸银负载硅胶层析柱则可实现极性相似的饱和化合物和不饱和化合物的分离[30 ,49 ] ;尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
... ,49 ];尿素缩合或者分子筛被用以分离直链化合物和非直链(支链或环状)化合物[30 ,49 ] .然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
... ,49 ].然而,自然环境样品经过以上湿化学萃取分离提纯后,只能得到一系列极性和化学结构相似的、含有目标生物标志物的组分集,之后还需要通过制备色谱技术才能获得目标生物标志物单体. ...
Towards radiocarbon dating of single foraminifera with a gas ion source
1
2013
... 天然样品的CSRA分析中,PCGC是最早发展起来的用于同位素分析的高纯度单体制备色谱技术.PCGC具有高的分离能力,克服了毛细管气相色谱的有限容量(<500 ng/compound,compound为单个化合物)的技术限制.整个装置由2部分组成:气相色谱和Gerstel八通阀回收装置,共6个回收窗口(图1 ).气相色谱端耦合了CIS系统程序升温的PTV进样口、大口径色谱柱(0.53 mm(内径,i, d),0.5 μm(固定相膜厚,film thickness))、FID检测器、十字分流阀.溶剂排空方式的PTV进样口和大孔径色谱柱的使用可实现大体积(≥5 μL/injection)、大容量(约2 μg/compound)进样,通过30~50次的重复进样可以收集足够量的目标化合物(60~100 μg/compound)满足现代高灵敏度AMS[50 ,51 ] 的14 C同位素测定需要.经过高效气相色谱柱分离的目标有机化合物单体通过十字分流阀,约1%进入FID检测器,其余99%通过毛细管传输线(transfer line,0.32 mm(内径,i.d.))进入Gerstel八通阀回收装置;低的保留时间漂移程度(标准偏差最大为0.03 min)保证目标化合物多次收集的准确性和可靠性[26 ] .Eglinton等[26 ] 首次应用PCGC分离富集了海洋沉积物中常见的类脂生物标志物单体,实现了自然环境浓度水平的单体14 C测定.然而CSRA技术仍然遇到很多的挑战和限制,导致其还没有真正实现全自动化,甚至相关的质量控制标准也未建立.在自然环境研究中,CSRA技术的应用所面临的挑战和瓶颈主要是环境样品中常见的几类生物标志物含量都较低,其有机化合物单体的纯化制备过程复杂繁琐且技术要求高.随着高灵敏度加速器质谱的发展,14 C同位素测定所需的样品量越来越小,甚至能够测试最低至几个微克碳的样品[36 ,52 ] .因此,过程空白或外源碳(Cex )对样品尤其是小样品最终14 C结果产生的影响就愈加明显[37 ,40 ,53 ] . ...
A versatile gas interface for routine radiocarbon analysis with a gas ion source
1
2013
... 天然样品的CSRA分析中,PCGC是最早发展起来的用于同位素分析的高纯度单体制备色谱技术.PCGC具有高的分离能力,克服了毛细管气相色谱的有限容量(<500 ng/compound,compound为单个化合物)的技术限制.整个装置由2部分组成:气相色谱和Gerstel八通阀回收装置,共6个回收窗口(图1 ).气相色谱端耦合了CIS系统程序升温的PTV进样口、大口径色谱柱(0.53 mm(内径,i, d),0.5 μm(固定相膜厚,film thickness))、FID检测器、十字分流阀.溶剂排空方式的PTV进样口和大孔径色谱柱的使用可实现大体积(≥5 μL/injection)、大容量(约2 μg/compound)进样,通过30~50次的重复进样可以收集足够量的目标化合物(60~100 μg/compound)满足现代高灵敏度AMS[50 ,51 ] 的14 C同位素测定需要.经过高效气相色谱柱分离的目标有机化合物单体通过十字分流阀,约1%进入FID检测器,其余99%通过毛细管传输线(transfer line,0.32 mm(内径,i.d.))进入Gerstel八通阀回收装置;低的保留时间漂移程度(标准偏差最大为0.03 min)保证目标化合物多次收集的准确性和可靠性[26 ] .Eglinton等[26 ] 首次应用PCGC分离富集了海洋沉积物中常见的类脂生物标志物单体,实现了自然环境浓度水平的单体14 C测定.然而CSRA技术仍然遇到很多的挑战和限制,导致其还没有真正实现全自动化,甚至相关的质量控制标准也未建立.在自然环境研究中,CSRA技术的应用所面临的挑战和瓶颈主要是环境样品中常见的几类生物标志物含量都较低,其有机化合物单体的纯化制备过程复杂繁琐且技术要求高.随着高灵敏度加速器质谱的发展,14 C同位素测定所需的样品量越来越小,甚至能够测试最低至几个微克碳的样品[36 ,52 ] .因此,过程空白或外源碳(Cex )对样品尤其是小样品最终14 C结果产生的影响就愈加明显[37 ,40 ,53 ] . ...
Developments in micro-sample 14 C AMS at the ANTARES AMS facility
1
2010
... 天然样品的CSRA分析中,PCGC是最早发展起来的用于同位素分析的高纯度单体制备色谱技术.PCGC具有高的分离能力,克服了毛细管气相色谱的有限容量(<500 ng/compound,compound为单个化合物)的技术限制.整个装置由2部分组成:气相色谱和Gerstel八通阀回收装置,共6个回收窗口(图1 ).气相色谱端耦合了CIS系统程序升温的PTV进样口、大口径色谱柱(0.53 mm(内径,i, d),0.5 μm(固定相膜厚,film thickness))、FID检测器、十字分流阀.溶剂排空方式的PTV进样口和大孔径色谱柱的使用可实现大体积(≥5 μL/injection)、大容量(约2 μg/compound)进样,通过30~50次的重复进样可以收集足够量的目标化合物(60~100 μg/compound)满足现代高灵敏度AMS[50 ,51 ] 的14 C同位素测定需要.经过高效气相色谱柱分离的目标有机化合物单体通过十字分流阀,约1%进入FID检测器,其余99%通过毛细管传输线(transfer line,0.32 mm(内径,i.d.))进入Gerstel八通阀回收装置;低的保留时间漂移程度(标准偏差最大为0.03 min)保证目标化合物多次收集的准确性和可靠性[26 ] .Eglinton等[26 ] 首次应用PCGC分离富集了海洋沉积物中常见的类脂生物标志物单体,实现了自然环境浓度水平的单体14 C测定.然而CSRA技术仍然遇到很多的挑战和限制,导致其还没有真正实现全自动化,甚至相关的质量控制标准也未建立.在自然环境研究中,CSRA技术的应用所面临的挑战和瓶颈主要是环境样品中常见的几类生物标志物含量都较低,其有机化合物单体的纯化制备过程复杂繁琐且技术要求高.随着高灵敏度加速器质谱的发展,14 C同位素测定所需的样品量越来越小,甚至能够测试最低至几个微克碳的样品[36 ,52 ] .因此,过程空白或外源碳(Cex )对样品尤其是小样品最终14 C结果产生的影响就愈加明显[37 ,40 ,53 ] . ...
Compound-specific radiocarbon analysis—Analytical challenges and applications
2
2009
... 天然样品的CSRA分析中,PCGC是最早发展起来的用于同位素分析的高纯度单体制备色谱技术.PCGC具有高的分离能力,克服了毛细管气相色谱的有限容量(<500 ng/compound,compound为单个化合物)的技术限制.整个装置由2部分组成:气相色谱和Gerstel八通阀回收装置,共6个回收窗口(图1 ).气相色谱端耦合了CIS系统程序升温的PTV进样口、大口径色谱柱(0.53 mm(内径,i, d),0.5 μm(固定相膜厚,film thickness))、FID检测器、十字分流阀.溶剂排空方式的PTV进样口和大孔径色谱柱的使用可实现大体积(≥5 μL/injection)、大容量(约2 μg/compound)进样,通过30~50次的重复进样可以收集足够量的目标化合物(60~100 μg/compound)满足现代高灵敏度AMS[50 ,51 ] 的14 C同位素测定需要.经过高效气相色谱柱分离的目标有机化合物单体通过十字分流阀,约1%进入FID检测器,其余99%通过毛细管传输线(transfer line,0.32 mm(内径,i.d.))进入Gerstel八通阀回收装置;低的保留时间漂移程度(标准偏差最大为0.03 min)保证目标化合物多次收集的准确性和可靠性[26 ] .Eglinton等[26 ] 首次应用PCGC分离富集了海洋沉积物中常见的类脂生物标志物单体,实现了自然环境浓度水平的单体14 C测定.然而CSRA技术仍然遇到很多的挑战和限制,导致其还没有真正实现全自动化,甚至相关的质量控制标准也未建立.在自然环境研究中,CSRA技术的应用所面临的挑战和瓶颈主要是环境样品中常见的几类生物标志物含量都较低,其有机化合物单体的纯化制备过程复杂繁琐且技术要求高.随着高灵敏度加速器质谱的发展,14 C同位素测定所需的样品量越来越小,甚至能够测试最低至几个微克碳的样品[36 ,52 ] .因此,过程空白或外源碳(Cex )对样品尤其是小样品最终14 C结果产生的影响就愈加明显[37 ,40 ,53 ] . ...
... 自从Eglinton等[26 ] 首次将单体分子放射性碳同位素分析技术应用于生物标志物14 C研究以来,该技术发展迅速,越来越多地应用于海洋科学、环境科学、生物地球化学和古气候学等领域,尤其是研究碳源和碳循环有关的问题.并且,随着高灵敏度AMS的快速发展,已经实现了超微样品14 C浓度的测试,甚至于1 μg C的测试[40 ] .但是,目前CSRA技术发展的最大限制是如何从复杂的自然环境样品中分离和收集纯的、大量的单体分子化合物,同时减少CSRA过程中引入的外源碳的污染[37 ,53 ] .近些年来,不断改进的PCGC和HPLC技术极大地扩展了生物标志物的应用,同时也提高了目标化合物分离的纯度和回收效率.目前,利用PCGC和Prep-HPLC等制备色谱进行分离和富集生物标志物单体时,对过程空白贡献的外源碳是1~2 μg C,因此,为了确保干扰物质低于10%,至少需要富集10~20 μg C以上的生物标志物单体进行其14 C测试.但据已有研究指出,CSRA技术过程空白具有高度的时空不确定性,所以将来的研究将需要更加关注“零外源碳”的分离纯化过程和严格的质量控制. ...
Evaluation of gas chromatographic isotope fractionation and process contamination by carbon in compound-specific radiocarbon analysis
6
2007
... 首先,保证一个无14 C-标记的实验环境是进行自然水平14 C浓度水平分析测试的前提.因为即使很少的14 C-标记化合物,也足以对放射性碳同位素分析造成非常大的偏差. 14 C/12 C的自然含量仅为1.2×10-12 ,即使3 × 10-18 ng的14 C也可以使25 μg C样品的Δ14 C值发生很大的改变(从+100‰到+1100‰)[54 ,55 ] ,并且14 C-标记的化合物不能通过GC-FID等技术手段进行快速的监测.不过,尽管14 C-标记化合物是14 C分析中最为严重的外源碳污染,但是严格的实验室管理措施可以避免添加14 C的污染[55 ] . ...
... 定量估算PCGC分离过程所引入外源碳的方法分别为直接法(directly method)和间接法(indirectly method)[38 ,57 ] .直接法是假定在PCGC分离过程中的外源碳主要是来自柱流失(包括过去样品的残留和/或GC色谱柱固定相的脱落),且柱流失不随时间和温度的变化而改变.在无溶剂条件下运行PCGC,馏分收集器按照目标化合物的截留时间收集相同条件下的仪器本底空白.Ziolkowski等[38 ] 通过直接法评估了PCGC分离过程中空白的质量随馏分收集器的收集窗口时间增加而增加,平均产生外源碳为(0.1±0.05) μg C /min(Fm=0.125±0.034).而间接方法则是通过加入已知14 C同位素水平的参考标准——“现代源(modern)”有机化合物标准(Fm=1)和“化石源(fossil)”有机化合物标准(Fm=0),分别评估PCGC分离过程中引入的化石源空白碳和现代空白碳的质量.二者质量加和即为引入的外源碳空白总质量,二者Fm的质量加权平均值即为外源碳空白的同位素特征值[38 ] .Ziolkowski等[38 ] 的研究结果显示通过间接法评估得到的PCGC分离过程中引入的外源碳中化石源空白碳和现代源空白碳的贡献分别是Cpcgc-fossil =(0.4±0.2) μg C/min和Cpcgc-modern =(0.2±0.1) μg C/min.因此,应用间接方法估算的外源碳的贡献是Cpcgc =(0.6±0.3) μg C/min(Fm=0.3±0.1).2种方法估算的PCGC分离过程中引入外源碳的贡献相差较大(0.5 μg C),这可能是在有溶剂条件下,来自色谱柱的外源碳更容易被洗脱以及样品的记忆效应和/或进样口的污染可能也是间接法呈现较高过程空白的影响因素.但Coppola等[57 ] 的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素.Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... 另外,稳定碳同位素手段也可以用来初步鉴别CSRA是否受到明显外来碳的影响.假定外源碳和目标化合物的13 C值有明显的差别,通过比较稳定碳同位素比质谱仪测定的目标有机物单体δ13 C值和PCGC分离富集回收后的目标化合物的δ13 C值之间的差异,也可以评估是否存在有机溶剂残留或者是其他污染外源碳被引入[26 ,54 ] . ...
... 由于PCGC分离过程中目标化合物在高温条件下以气态的形式传输后进入捕集管,整个系统气密性的可持续性决定了目标化合物回收率的好坏,实际实验过程中PCGC的回收率一般在70%~80%[26 ,60 ] .在色谱分离过程中通常会发生同位素分馏,随着色谱信号的流出,其碳同位素组成是不断变化的[61 ,62 ] .因此,PCGC分离富集过程中目标化合物不完全回收往往也会导致一定的碳同位素分馏.Zencak等[54 ] 测定了香草醛标准不同截留时间馏分的δ13 C,结果显示其谱峰的前沿部分(-15.75‰)明显偏正于峰尾部分(-49.91‰),这与Eglinton等[26 ] 的结果一致.碳同位素呈现逆同位素效应(inverse isotope effect),即重的碳同位素先流出而轻的同位素后流出,这与Cl等同位素效应相反[63 ] .因此当目标化合物不完全分离或收集,会发生较为明显的碳同位素分馏.Zencak等[54 ] 同时指出,未经处理的香草醛δ13 C比值(-31.6‰)与经PCGC完整收集的香草醛δ13 C比值(-31.3‰)一致.但实际中,尤其是处理海洋沉积物等基质复杂的样品,为了获取高纯度的目标化合物,通常会缩短组分截留窗口的时间,即靠近目标化合物拖尾或前沿的部分截留,这可能导致目标化合物不完全回收而引起一定程度的碳同位素分馏.理论上,δ14 C的同位素分馏应当是δ13 C分馏的2倍,呈现和13 C同样的逆同位素效应.但是,放射性碳数据通常是报道经过δ13 C=-25‰校正后的Fm和Δ14 C值[64 ] .因此,即使发生碳同位素分馏,只会影响目标化合物的回收率及δ13 C数据的准确性,而不会影响最终报道的放射性碳同位素数据[64 ] . ...
... [54 ]同时指出,未经处理的香草醛δ13 C比值(-31.6‰)与经PCGC完整收集的香草醛δ13 C比值(-31.3‰)一致.但实际中,尤其是处理海洋沉积物等基质复杂的样品,为了获取高纯度的目标化合物,通常会缩短组分截留窗口的时间,即靠近目标化合物拖尾或前沿的部分截留,这可能导致目标化合物不完全回收而引起一定程度的碳同位素分馏.理论上,δ14 C的同位素分馏应当是δ13 C分馏的2倍,呈现和13 C同样的逆同位素效应.但是,放射性碳数据通常是报道经过δ13 C=-25‰校正后的Fm和Δ14 C值[64 ] .因此,即使发生碳同位素分馏,只会影响目标化合物的回收率及δ13 C数据的准确性,而不会影响最终报道的放射性碳同位素数据[64 ] . ...
... 传统的PCGC配置了大孔径色谱柱(0.53 mm i.d.),提供了大负载容量的同时,牺牲了色谱的分离能力[43 ] .而为了弥补传统PCGC分离能力且减少或避免使用可能引入外源碳的其他湿化学萃取分离过程[38 ,54 ,57 ] ,有报道指出可以通过串联色谱及多维色谱技术提高传统PCGC对复杂基质的分离能力,同时也减少了繁琐的化学分离前处理过程中潜在外源碳的引入[42 ] .Ball等[42 ] 将二维PCGC应用于CSRA样品的分离.与传统PCGC相比,其增加了一个中心切割微流体切换装置(Deans pneumatic microfluidic switching device)以及串联2个不同固定相的色谱柱(Rtx-5 MS, 0.50 μm df和DB-17 MS, 0.25 μm df).尽管增加了微流体切换装置及色谱柱数量,但其保留时间漂移程度很小(标准偏差最大为0.016 min),这与传统PCGC的性能基本一致[26 ] .而多次进样发现其FID峰面积的偏差为1.1%~1.2%[42 ] ,这可能是与PTV进样模式所引起的扰动有关.传统PCGC柱流失对过程空白的贡献是(0.6±0.3) μg C/min,但如果无溶剂空运行PCGC,则柱流失仅贡献了0.1 μg C/min[38 ] .而在二维PCGC中,2°色谱柱仅仅从1°柱中接收有限量的样品,而并没有溶剂进入2°柱,因此柱流失并不是二维PCGC技术外源碳的重要来源.另外,在二维PCGC中使用了较薄的(0.25 μm, df)和低柱流失的固定相,可以进一步降低柱流失的贡献.Ball等[42 ] 通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定. ...
Tips and traps in the 14 C bio-AMS preparation laboratory
2
2000
... 首先,保证一个无14 C-标记的实验环境是进行自然水平14 C浓度水平分析测试的前提.因为即使很少的14 C-标记化合物,也足以对放射性碳同位素分析造成非常大的偏差. 14 C/12 C的自然含量仅为1.2×10-12 ,即使3 × 10-18 ng的14 C也可以使25 μg C样品的Δ14 C值发生很大的改变(从+100‰到+1100‰)[54 ,55 ] ,并且14 C-标记的化合物不能通过GC-FID等技术手段进行快速的监测.不过,尽管14 C-标记化合物是14 C分析中最为严重的外源碳污染,但是严格的实验室管理措施可以避免添加14 C的污染[55 ] . ...
... [55 ]. ...
Quantifying archaeal community autotrophy in the mesopelagic ocean using natural radiocarbon
1
2006
... 随着CSRA技术在自然环境样品中的应用,样品量逐渐向超微样品(<25 μg C)发展,此时过程空白也就成为了CSRA样品14 C测试精确度和准确度的实际限制因素[37 ,56 ] .自然环境中的样品,化学萃取和PCGC分离富集过程中引入的外源碳是过程空白的主要贡献,远远高于在真空线转换为CO2 和/或石墨化过程中外源碳的量;并且当样品量变小时(<100 μg C),过程空白的评价显得更为重要[40 ] .在PCGC分离过程中,色谱柱流失是外源碳的主要来源之一.Eglinton等[26 ] 发现,当PCGC使用高容量色谱柱(>1 μm固定相膜厚)时,尤其是高沸点的有机化合物单体组分中,GC-FID能够明显从PCGC-U型捕集管中检测出聚甲基硅酮的降解产物;而对于使用较薄的固定相时(≤0.5 μm),色谱柱流失的影响则明显减少. ...
Druffel E R M. Extraneous carbon assessments in radiocarbon measurements of black carbon in environmental matrices
5
2013
... 定量估算PCGC分离过程所引入外源碳的方法分别为直接法(directly method)和间接法(indirectly method)[38 ,57 ] .直接法是假定在PCGC分离过程中的外源碳主要是来自柱流失(包括过去样品的残留和/或GC色谱柱固定相的脱落),且柱流失不随时间和温度的变化而改变.在无溶剂条件下运行PCGC,馏分收集器按照目标化合物的截留时间收集相同条件下的仪器本底空白.Ziolkowski等[38 ] 通过直接法评估了PCGC分离过程中空白的质量随馏分收集器的收集窗口时间增加而增加,平均产生外源碳为(0.1±0.05) μg C /min(Fm=0.125±0.034).而间接方法则是通过加入已知14 C同位素水平的参考标准——“现代源(modern)”有机化合物标准(Fm=1)和“化石源(fossil)”有机化合物标准(Fm=0),分别评估PCGC分离过程中引入的化石源空白碳和现代空白碳的质量.二者质量加和即为引入的外源碳空白总质量,二者Fm的质量加权平均值即为外源碳空白的同位素特征值[38 ] .Ziolkowski等[38 ] 的研究结果显示通过间接法评估得到的PCGC分离过程中引入的外源碳中化石源空白碳和现代源空白碳的贡献分别是Cpcgc-fossil =(0.4±0.2) μg C/min和Cpcgc-modern =(0.2±0.1) μg C/min.因此,应用间接方法估算的外源碳的贡献是Cpcgc =(0.6±0.3) μg C/min(Fm=0.3±0.1).2种方法估算的PCGC分离过程中引入外源碳的贡献相差较大(0.5 μg C),这可能是在有溶剂条件下,来自色谱柱的外源碳更容易被洗脱以及样品的记忆效应和/或进样口的污染可能也是间接法呈现较高过程空白的影响因素.但Coppola等[57 ] 的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素.Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... [57 ]的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素.Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... [57 ]的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... [57 ],这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
... 传统的PCGC配置了大孔径色谱柱(0.53 mm i.d.),提供了大负载容量的同时,牺牲了色谱的分离能力[43 ] .而为了弥补传统PCGC分离能力且减少或避免使用可能引入外源碳的其他湿化学萃取分离过程[38 ,54 ,57 ] ,有报道指出可以通过串联色谱及多维色谱技术提高传统PCGC对复杂基质的分离能力,同时也减少了繁琐的化学分离前处理过程中潜在外源碳的引入[42 ] .Ball等[42 ] 将二维PCGC应用于CSRA样品的分离.与传统PCGC相比,其增加了一个中心切割微流体切换装置(Deans pneumatic microfluidic switching device)以及串联2个不同固定相的色谱柱(Rtx-5 MS, 0.50 μm df和DB-17 MS, 0.25 μm df).尽管增加了微流体切换装置及色谱柱数量,但其保留时间漂移程度很小(标准偏差最大为0.016 min),这与传统PCGC的性能基本一致[26 ] .而多次进样发现其FID峰面积的偏差为1.1%~1.2%[42 ] ,这可能是与PTV进样模式所引起的扰动有关.传统PCGC柱流失对过程空白的贡献是(0.6±0.3) μg C/min,但如果无溶剂空运行PCGC,则柱流失仅贡献了0.1 μg C/min[38 ] .而在二维PCGC中,2°色谱柱仅仅从1°柱中接收有限量的样品,而并没有溶剂进入2°柱,因此柱流失并不是二维PCGC技术外源碳的重要来源.另外,在二维PCGC中使用了较薄的(0.25 μm, df)和低柱流失的固定相,可以进一步降低柱流失的贡献.Ball等[42 ] 通过对香草酸甲酯等6个已知14 C同位素水平的有机化合物标准分离前后的Δ14 C值进行比较,其Δ(Δ14 C isolated - Δ14 Cinitial )的差值均小于等于(6.7±5.0)‰,这一差异小于AMS放射性碳同位素的测量误差(约15‰)[30 ] ,低的过程空白的贡献说明二维PCGC分离技术更适合于超微样品的CSRA测定. ...
黄河颗粒态及渤、黄海现代沉积有机质的组成和同位素分布特征及源项解析[D]
1
2014
... 定量估算PCGC分离过程所引入外源碳的方法分别为直接法(directly method)和间接法(indirectly method)[38 ,57 ] .直接法是假定在PCGC分离过程中的外源碳主要是来自柱流失(包括过去样品的残留和/或GC色谱柱固定相的脱落),且柱流失不随时间和温度的变化而改变.在无溶剂条件下运行PCGC,馏分收集器按照目标化合物的截留时间收集相同条件下的仪器本底空白.Ziolkowski等[38 ] 通过直接法评估了PCGC分离过程中空白的质量随馏分收集器的收集窗口时间增加而增加,平均产生外源碳为(0.1±0.05) μg C /min(Fm=0.125±0.034).而间接方法则是通过加入已知14 C同位素水平的参考标准——“现代源(modern)”有机化合物标准(Fm=1)和“化石源(fossil)”有机化合物标准(Fm=0),分别评估PCGC分离过程中引入的化石源空白碳和现代空白碳的质量.二者质量加和即为引入的外源碳空白总质量,二者Fm的质量加权平均值即为外源碳空白的同位素特征值[38 ] .Ziolkowski等[38 ] 的研究结果显示通过间接法评估得到的PCGC分离过程中引入的外源碳中化石源空白碳和现代源空白碳的贡献分别是Cpcgc-fossil =(0.4±0.2) μg C/min和Cpcgc-modern =(0.2±0.1) μg C/min.因此,应用间接方法估算的外源碳的贡献是Cpcgc =(0.6±0.3) μg C/min(Fm=0.3±0.1).2种方法估算的PCGC分离过程中引入外源碳的贡献相差较大(0.5 μg C),这可能是在有溶剂条件下,来自色谱柱的外源碳更容易被洗脱以及样品的记忆效应和/或进样口的污染可能也是间接法呈现较高过程空白的影响因素.但Coppola等[57 ] 的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素.Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
黄河颗粒态及渤、黄海现代沉积有机质的组成和同位素分布特征及源项解析[D]
1
2014
... 定量估算PCGC分离过程所引入外源碳的方法分别为直接法(directly method)和间接法(indirectly method)[38 ,57 ] .直接法是假定在PCGC分离过程中的外源碳主要是来自柱流失(包括过去样品的残留和/或GC色谱柱固定相的脱落),且柱流失不随时间和温度的变化而改变.在无溶剂条件下运行PCGC,馏分收集器按照目标化合物的截留时间收集相同条件下的仪器本底空白.Ziolkowski等[38 ] 通过直接法评估了PCGC分离过程中空白的质量随馏分收集器的收集窗口时间增加而增加,平均产生外源碳为(0.1±0.05) μg C /min(Fm=0.125±0.034).而间接方法则是通过加入已知14 C同位素水平的参考标准——“现代源(modern)”有机化合物标准(Fm=1)和“化石源(fossil)”有机化合物标准(Fm=0),分别评估PCGC分离过程中引入的化石源空白碳和现代空白碳的质量.二者质量加和即为引入的外源碳空白总质量,二者Fm的质量加权平均值即为外源碳空白的同位素特征值[38 ] .Ziolkowski等[38 ] 的研究结果显示通过间接法评估得到的PCGC分离过程中引入的外源碳中化石源空白碳和现代源空白碳的贡献分别是Cpcgc-fossil =(0.4±0.2) μg C/min和Cpcgc-modern =(0.2±0.1) μg C/min.因此,应用间接方法估算的外源碳的贡献是Cpcgc =(0.6±0.3) μg C/min(Fm=0.3±0.1).2种方法估算的PCGC分离过程中引入外源碳的贡献相差较大(0.5 μg C),这可能是在有溶剂条件下,来自色谱柱的外源碳更容易被洗脱以及样品的记忆效应和/或进样口的污染可能也是间接法呈现较高过程空白的影响因素.但Coppola等[57 ] 的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素.Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
Ultra small-mass AMS 14 C sample preparation and analyses at KCCAMS/UCI Facility
1
2007
... 定量估算PCGC分离过程所引入外源碳的方法分别为直接法(directly method)和间接法(indirectly method)[38 ,57 ] .直接法是假定在PCGC分离过程中的外源碳主要是来自柱流失(包括过去样品的残留和/或GC色谱柱固定相的脱落),且柱流失不随时间和温度的变化而改变.在无溶剂条件下运行PCGC,馏分收集器按照目标化合物的截留时间收集相同条件下的仪器本底空白.Ziolkowski等[38 ] 通过直接法评估了PCGC分离过程中空白的质量随馏分收集器的收集窗口时间增加而增加,平均产生外源碳为(0.1±0.05) μg C /min(Fm=0.125±0.034).而间接方法则是通过加入已知14 C同位素水平的参考标准——“现代源(modern)”有机化合物标准(Fm=1)和“化石源(fossil)”有机化合物标准(Fm=0),分别评估PCGC分离过程中引入的化石源空白碳和现代空白碳的质量.二者质量加和即为引入的外源碳空白总质量,二者Fm的质量加权平均值即为外源碳空白的同位素特征值[38 ] .Ziolkowski等[38 ] 的研究结果显示通过间接法评估得到的PCGC分离过程中引入的外源碳中化石源空白碳和现代源空白碳的贡献分别是Cpcgc-fossil =(0.4±0.2) μg C/min和Cpcgc-modern =(0.2±0.1) μg C/min.因此,应用间接方法估算的外源碳的贡献是Cpcgc =(0.6±0.3) μg C/min(Fm=0.3±0.1).2种方法估算的PCGC分离过程中引入外源碳的贡献相差较大(0.5 μg C),这可能是在有溶剂条件下,来自色谱柱的外源碳更容易被洗脱以及样品的记忆效应和/或进样口的污染可能也是间接法呈现较高过程空白的影响因素.但Coppola等[57 ] 的结果显示PCGC分离过程即使使用了色谱级溶剂,通过直接法估算的外源碳是(0.1±0.1) μg C/min,认为PCGC分离过程中溶剂并不是高外源碳的主要因素.Ziolkowski等[38 ] 的结果显示,2种方法外源碳的Fm值分别为0.125和0.3,表明PCGC过程中引入的外源碳主要是“化石源”碳的贡献.由于色谱柱固定相主要是石油产品,因此色谱柱流失可能是PCGC过程中外源碳中老碳的主要来源.但我们的结果显示,PCGC过程产生约1.3 μg C外源碳(表1 ),直接法和间接法引入的外源碳Fm值分别为0.6224和0.6793,这表明了色谱柱柱流失的贡献相对较小.另外,PCGC分离富集后使用有机溶剂(比如CH2 Cl2 )将目标化合物从PCGC捕集管转移至真空线上的石英管中待进行CO2 的转化,石英管内有机溶剂的不完全移除可能也是外源碳中老碳的一个重要来源.由于实验过程大多使用低沸点、易挥发的有机溶剂,所以无法使用GC-FID等来监测未完全移除的有机溶剂的量,导致难以定量估算残余溶剂对过程空白的贡献.前人将PCGC收集后的目标有机化合物,通过对比高效气相色谱的定量结果和真空线燃烧后的CO2 产量之间的差异来初步评估溶剂残留的影响,发现两者大致呈较好的线性关系,但某些样品中也会出现不一致的情况.这可能是因为某些单体化合物样品中有机溶剂未被完全移除或者是移除的程度不一致[26 ] .通过燃烧办法计算的低沸点或易挥发化合物的产量容易被低估,因为在氮吹或者旋转蒸发过程中这些化合物容易挥发损失.对于高沸点有机化合物,如碳数大于20的烷烃,则明显呈现高的燃烧产量,这主要由于在氮吹过程中这些化合物在有机溶剂表面可以形成黏性的皮肤,从而导致有机溶剂不完全去除.另外,商品化的有机溶剂中可能含有稳定剂(如2,6-二叔丁基对甲酚),这也是引入化石源碳污染途径之一[54 ] .Ziolkowski等[38 ] 同时也评估了湿化学分离过程引入的外源碳(Cchemistry ),结果显示其和PCGC过程引入的外源碳的贡献相当(表1 ),但Coppola等[57 ] 的结果则显示相对湿化学分离过程,PCGC过程引入外源碳的相对比例较小(表1 ),仅占过程空白(Cchemistry+pcgc )的10%左右,而Cchemistry 贡献相对高可能是来自阳离子交换柱分离过程.通过比较同一实验室不同时间(2012年和2011年)评估的过程空白(表1 ),显示尽管在同样的实验室,外源碳(Cex )对CSRA过程空白的贡献变化很大[57 ] ,这与某些实验室过程空白的贡献较为稳定不一致[59 ] .陶舒琴[58 ] 利用直接法和间接法评估CSRA前处理过程(Cchemistry+pcgc )中产生的过程空白外源碳的贡献分别是Cchemistry+pcgc =(2.44±0.22) μg C/min(Fm = 0.62±0.01)和Cchemistry+pcgc =(1.25±0.24) μg C/min(Fm= 0.68±0.13).由此可知,CSRA的过程空白具有较高的时空不确定性,因此实现好的CSRA质控必须定期对过程空白进行评估. ...
Biogeochemical Applications of Compound-Specific Radiocarbon Analysis[R].
1
1999
... 由于PCGC分离过程中目标化合物在高温条件下以气态的形式传输后进入捕集管,整个系统气密性的可持续性决定了目标化合物回收率的好坏,实际实验过程中PCGC的回收率一般在70%~80%[26 ,60 ] .在色谱分离过程中通常会发生同位素分馏,随着色谱信号的流出,其碳同位素组成是不断变化的[61 ,62 ] .因此,PCGC分离富集过程中目标化合物不完全回收往往也会导致一定的碳同位素分馏.Zencak等[54 ] 测定了香草醛标准不同截留时间馏分的δ13 C,结果显示其谱峰的前沿部分(-15.75‰)明显偏正于峰尾部分(-49.91‰),这与Eglinton等[26 ] 的结果一致.碳同位素呈现逆同位素效应(inverse isotope effect),即重的碳同位素先流出而轻的同位素后流出,这与Cl等同位素效应相反[63 ] .因此当目标化合物不完全分离或收集,会发生较为明显的碳同位素分馏.Zencak等[54 ] 同时指出,未经处理的香草醛δ13 C比值(-31.6‰)与经PCGC完整收集的香草醛δ13 C比值(-31.3‰)一致.但实际中,尤其是处理海洋沉积物等基质复杂的样品,为了获取高纯度的目标化合物,通常会缩短组分截留窗口的时间,即靠近目标化合物拖尾或前沿的部分截留,这可能导致目标化合物不完全回收而引起一定程度的碳同位素分馏.理论上,δ14 C的同位素分馏应当是δ13 C分馏的2倍,呈现和13 C同样的逆同位素效应.但是,放射性碳数据通常是报道经过δ13 C=-25‰校正后的Fm和Δ14 C值[64 ] .因此,即使发生碳同位素分馏,只会影响目标化合物的回收率及δ13 C数据的准确性,而不会影响最终报道的放射性碳同位素数据[64 ] . ...
Compound-specific isotopic analyses: A novel tool for reconstruction of ancient biogeochemical processes
1
1990
... 由于PCGC分离过程中目标化合物在高温条件下以气态的形式传输后进入捕集管,整个系统气密性的可持续性决定了目标化合物回收率的好坏,实际实验过程中PCGC的回收率一般在70%~80%[26 ,60 ] .在色谱分离过程中通常会发生同位素分馏,随着色谱信号的流出,其碳同位素组成是不断变化的[61 ,62 ] .因此,PCGC分离富集过程中目标化合物不完全回收往往也会导致一定的碳同位素分馏.Zencak等[54 ] 测定了香草醛标准不同截留时间馏分的δ13 C,结果显示其谱峰的前沿部分(-15.75‰)明显偏正于峰尾部分(-49.91‰),这与Eglinton等[26 ] 的结果一致.碳同位素呈现逆同位素效应(inverse isotope effect),即重的碳同位素先流出而轻的同位素后流出,这与Cl等同位素效应相反[63 ] .因此当目标化合物不完全分离或收集,会发生较为明显的碳同位素分馏.Zencak等[54 ] 同时指出,未经处理的香草醛δ13 C比值(-31.6‰)与经PCGC完整收集的香草醛δ13 C比值(-31.3‰)一致.但实际中,尤其是处理海洋沉积物等基质复杂的样品,为了获取高纯度的目标化合物,通常会缩短组分截留窗口的时间,即靠近目标化合物拖尾或前沿的部分截留,这可能导致目标化合物不完全回收而引起一定程度的碳同位素分馏.理论上,δ14 C的同位素分馏应当是δ13 C分馏的2倍,呈现和13 C同样的逆同位素效应.但是,放射性碳数据通常是报道经过δ13 C=-25‰校正后的Fm和Δ14 C值[64 ] .因此,即使发生碳同位素分馏,只会影响目标化合物的回收率及δ13 C数据的准确性,而不会影响最终报道的放射性碳同位素数据[64 ] . ...
Quantitative evaluation of carbon isotopic fractionation during reversed-phase high-performance liquid chromatography
1
1997
... 由于PCGC分离过程中目标化合物在高温条件下以气态的形式传输后进入捕集管,整个系统气密性的可持续性决定了目标化合物回收率的好坏,实际实验过程中PCGC的回收率一般在70%~80%[26 ,60 ] .在色谱分离过程中通常会发生同位素分馏,随着色谱信号的流出,其碳同位素组成是不断变化的[61 ,62 ] .因此,PCGC分离富集过程中目标化合物不完全回收往往也会导致一定的碳同位素分馏.Zencak等[54 ] 测定了香草醛标准不同截留时间馏分的δ13 C,结果显示其谱峰的前沿部分(-15.75‰)明显偏正于峰尾部分(-49.91‰),这与Eglinton等[26 ] 的结果一致.碳同位素呈现逆同位素效应(inverse isotope effect),即重的碳同位素先流出而轻的同位素后流出,这与Cl等同位素效应相反[63 ] .因此当目标化合物不完全分离或收集,会发生较为明显的碳同位素分馏.Zencak等[54 ] 同时指出,未经处理的香草醛δ13 C比值(-31.6‰)与经PCGC完整收集的香草醛δ13 C比值(-31.3‰)一致.但实际中,尤其是处理海洋沉积物等基质复杂的样品,为了获取高纯度的目标化合物,通常会缩短组分截留窗口的时间,即靠近目标化合物拖尾或前沿的部分截留,这可能导致目标化合物不完全回收而引起一定程度的碳同位素分馏.理论上,δ14 C的同位素分馏应当是δ13 C分馏的2倍,呈现和13 C同样的逆同位素效应.但是,放射性碳数据通常是报道经过δ13 C=-25‰校正后的Fm和Δ14 C值[64 ] .因此,即使发生碳同位素分馏,只会影响目标化合物的回收率及δ13 C数据的准确性,而不会影响最终报道的放射性碳同位素数据[64 ] . ...
Chlorine isotope fractionation of a semi-volatile organochlorine compound during preparative megabore-column capillary gas chromatography
1
2006
... 由于PCGC分离过程中目标化合物在高温条件下以气态的形式传输后进入捕集管,整个系统气密性的可持续性决定了目标化合物回收率的好坏,实际实验过程中PCGC的回收率一般在70%~80%[26 ,60 ] .在色谱分离过程中通常会发生同位素分馏,随着色谱信号的流出,其碳同位素组成是不断变化的[61 ,62 ] .因此,PCGC分离富集过程中目标化合物不完全回收往往也会导致一定的碳同位素分馏.Zencak等[54 ] 测定了香草醛标准不同截留时间馏分的δ13 C,结果显示其谱峰的前沿部分(-15.75‰)明显偏正于峰尾部分(-49.91‰),这与Eglinton等[26 ] 的结果一致.碳同位素呈现逆同位素效应(inverse isotope effect),即重的碳同位素先流出而轻的同位素后流出,这与Cl等同位素效应相反[63 ] .因此当目标化合物不完全分离或收集,会发生较为明显的碳同位素分馏.Zencak等[54 ] 同时指出,未经处理的香草醛δ13 C比值(-31.6‰)与经PCGC完整收集的香草醛δ13 C比值(-31.3‰)一致.但实际中,尤其是处理海洋沉积物等基质复杂的样品,为了获取高纯度的目标化合物,通常会缩短组分截留窗口的时间,即靠近目标化合物拖尾或前沿的部分截留,这可能导致目标化合物不完全回收而引起一定程度的碳同位素分馏.理论上,δ14 C的同位素分馏应当是δ13 C分馏的2倍,呈现和13 C同样的逆同位素效应.但是,放射性碳数据通常是报道经过δ13 C=-25‰校正后的Fm和Δ14 C值[64 ] .因此,即使发生碳同位素分馏,只会影响目标化合物的回收率及δ13 C数据的准确性,而不会影响最终报道的放射性碳同位素数据[64 ] . ...
Radiocarbon-discussion reporting of 14 C data
2
1977
... 由于PCGC分离过程中目标化合物在高温条件下以气态的形式传输后进入捕集管,整个系统气密性的可持续性决定了目标化合物回收率的好坏,实际实验过程中PCGC的回收率一般在70%~80%[26 ,60 ] .在色谱分离过程中通常会发生同位素分馏,随着色谱信号的流出,其碳同位素组成是不断变化的[61 ,62 ] .因此,PCGC分离富集过程中目标化合物不完全回收往往也会导致一定的碳同位素分馏.Zencak等[54 ] 测定了香草醛标准不同截留时间馏分的δ13 C,结果显示其谱峰的前沿部分(-15.75‰)明显偏正于峰尾部分(-49.91‰),这与Eglinton等[26 ] 的结果一致.碳同位素呈现逆同位素效应(inverse isotope effect),即重的碳同位素先流出而轻的同位素后流出,这与Cl等同位素效应相反[63 ] .因此当目标化合物不完全分离或收集,会发生较为明显的碳同位素分馏.Zencak等[54 ] 同时指出,未经处理的香草醛δ13 C比值(-31.6‰)与经PCGC完整收集的香草醛δ13 C比值(-31.3‰)一致.但实际中,尤其是处理海洋沉积物等基质复杂的样品,为了获取高纯度的目标化合物,通常会缩短组分截留窗口的时间,即靠近目标化合物拖尾或前沿的部分截留,这可能导致目标化合物不完全回收而引起一定程度的碳同位素分馏.理论上,δ14 C的同位素分馏应当是δ13 C分馏的2倍,呈现和13 C同样的逆同位素效应.但是,放射性碳数据通常是报道经过δ13 C=-25‰校正后的Fm和Δ14 C值[64 ] .因此,即使发生碳同位素分馏,只会影响目标化合物的回收率及δ13 C数据的准确性,而不会影响最终报道的放射性碳同位素数据[64 ] . ...
... [64 ]. ...
The radiocarbon content of individual lignin-derived phenols: Technique and initial results
3
2000
... Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段.Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致.可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标.Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右.但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
... [65 ,66 ],而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
... [65 ].为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
Origins of lipid biomarkers in Santa Monica Basin surface sediment: A case study using compound-specific Δ14 C analysis
1
2001
... Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段.Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致.可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标.Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右.但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
Optimisation of derivatisation procedures for the determination of δ13 C values of amino acids by gas chromatography/combustion/isotope ratio mass spectrometry
1
2007
... Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段.Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致.可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标.Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右.但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
Origins of archaeal tetraether lipids in sediments: Insights from radiocarbon analysis
1
2008
... Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段.Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致.可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标.Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右.但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
Rapid isolation of biomarkers for compound specific radiocarbon dating using high-performance liquid chromatography and flow injection analysis-atmospheric pressure chemical ionisation mass spectrometry
4
2002
... Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段.Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致.可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标.Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右.但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
... [69 ]、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
... 利用半制备型氨基柱和分析型氨基柱的2步分离的Prep-HPLC技术被成功用于分离海洋沉积物中的GDGTs、植醇及甾醇等生物标志物[69 ] .该Prep-HPLC系统耦合了流动注射分析技术、大气压电离质谱、HP-1100液相色谱(HPLC)和馏分收集装置.根据液相色谱柱分离后的目标化合物单体进入大气压电离质谱(APCI-MS)检测器分子离子信号峰的保留时间设定馏分收集器的截留时间.化学分离纯化后的极性类脂组分(见本文第2部分介绍)通过半制备型氨基柱(Econosphere, 250 mm×10 mm, 10 μm; Alltech Associates)进行初步的分离收集,然后使用分析型氨基柱(Econosphere, 250 mm×4.6 mm, 5 μm; Alltech Associates)进一步分离纯化.Smittenberg等[69 ] 通过GC,GCMSD,HPLC-MS和核磁共振(13 C-NMR和1 H-NMR)手段分别对以上Prep-HPLC分离富集所得的甾醇类和GDGT(GDGT-0和Crenarcheaol)类有机化合物单体进行了纯度检测,类脂有机化合物单体纯度高于95%,且没有其他明显的外源碳的污染.与PCGC相比较,具有相对高的回收效率(>89%).Prep-HPLC成功地分离自然环境样品中高极性、高沸点(>C30)生物标志物,扩充生物标志物单体14 C的应用范围.此外,Prep-HPLC具有更大的柱容量(高达30 μg/injection),相对于PCGC分离富集的时间大大缩短. ...
... [69 ]通过GC,GCMSD,HPLC-MS和核磁共振(13 C-NMR和1 H-NMR)手段分别对以上Prep-HPLC分离富集所得的甾醇类和GDGT(GDGT-0和Crenarcheaol)类有机化合物单体进行了纯度检测,类脂有机化合物单体纯度高于95%,且没有其他明显的外源碳的污染.与PCGC相比较,具有相对高的回收效率(>89%).Prep-HPLC成功地分离自然环境样品中高极性、高沸点(>C30)生物标志物,扩充生物标志物单体14 C的应用范围.此外,Prep-HPLC具有更大的柱容量(高达30 μg/injection),相对于PCGC分离富集的时间大大缩短. ...
Radiocarbon analysis of individual Amino Acids: Carbon blank quantification for a small-sample high-pressure liquid chromatography purification method
1
2016
... Pearson等[5 ] 利用PCGC从海洋沉积物中分离甲藻甾醇等甾醇,发现甲藻甾醇等Δ14 C值与海水DIC的Δ14 C一致,显示其来自海洋浮游植物,这为海洋有机碳汇源解析模型提供了精确的海洋源端元特征值,提供了一个示踪海源有机碳汇的有效手段.Kusch等[7 ] 测定了长链烯酮和浮游有孔虫的14 C,二者在沉积柱中年龄一致.可以类推,地质样品中海源生物标志物如浮游藻类甾醇可以用来作为潜在的定年替代指标.Mcnichol等[65 ] 利用PCGC从标准树木样品成功地分离了木质素酚类化合物,并准确测定其Δ14 C值,其与总有机碳Δ14 C值差20‰左右.但是,PCGC分离技术只能用于分离弱极性和低分子量的化合物,比如正构烷烃及衍生化后的甾醇和脂肪酸[65 ,66 ] ,而利用PCGC分离这一类甾醇和酚类生物标志物需要添加衍生化试剂以适用于气相色谱的分离,在14 C同位素数据处理过程中需要对衍生化试剂添加碳的扣除校正,会增加目标化合物的测试偏差[67 ] .同时,衍生化试剂也会引入一定的外源碳[65 ] .为了避免衍生化添加碳的影响,制备液相色谱技术(Prep-HPLC)被广泛应用于分离富集植物、海洋和湖泊沉积物中的木质素[14 ,30 ] 、br-GDGTs[48 ] 、GDGTs[68 ,69 ] 、甾醇[69 ] 、氨基酸[70 ] 等高极性、高沸点和大分子有机化合物,以适用于CSRA测试. ...
A novel proxy for terrestrial organic matter in sediments based on branched and isoprenoid tetraether lipids
1
2004
... Birhkolz等[48 ] 改进了已有的Prep-HPLC技术,成功分离富集出湖泊沉积物中br-GDGTs化合物单体.首先利用反相-HPLC(Agilent Eclipse Plus C18 column,4.6 mm×150 mm, 3.5 μm)将br-GDGTs和共流出组分分离;为了除去反相-HPLC柱分离过程中引入的共流出组分和进一步纯化,使用正相-HPLC柱(Alltech Prevail Cyano column, 2.1 mm×150 mm, 3 μm)消除可能来自流动相的外源碳(12 μg C/60 min).改进后的HPLC技术贡献的外源碳为 (0.6±0.2) μg C(其F14 C为0.5±0.2),与Hou等[14 ] 结果基本一致,其外源碳的贡献约为2 μg C(Fm值为0.30).基于br-GDGTs的BIT指标已被广泛用来估算海洋环境中土壤有机质的贡献比例[71 ~73 ] ,但有报道指出湖泊[74 ] 和海洋[75 ] 也会产生br-GDGTs.湖泊、海洋环境样品中br-GDGTs的14 C同位素特征可以为br-GDGTs土壤源指标的应用提供重要的信息.Birkholz等[48 ] 发现Lake Lusvatnet沉积物中的br-GDGTs呈现较老的14 C年龄.结合湖泊水体年轻的DOC特征[76 ] ,可明确判断该湖泊br-GDGTs来源于外源陈化土壤的输入. ...
Origin and distribution of terrestrial organic matter in the NW Mediterranean (Gulf of Lions): Exploring the newly developed BIT index
0
2006
Multiple proxy estimates of source and spatial variation in organic matter in surface sediments from the southern Yellow Sea
1
2014
... Birhkolz等[48 ] 改进了已有的Prep-HPLC技术,成功分离富集出湖泊沉积物中br-GDGTs化合物单体.首先利用反相-HPLC(Agilent Eclipse Plus C18 column,4.6 mm×150 mm, 3.5 μm)将br-GDGTs和共流出组分分离;为了除去反相-HPLC柱分离过程中引入的共流出组分和进一步纯化,使用正相-HPLC柱(Alltech Prevail Cyano column, 2.1 mm×150 mm, 3 μm)消除可能来自流动相的外源碳(12 μg C/60 min).改进后的HPLC技术贡献的外源碳为 (0.6±0.2) μg C(其F14 C为0.5±0.2),与Hou等[14 ] 结果基本一致,其外源碳的贡献约为2 μg C(Fm值为0.30).基于br-GDGTs的BIT指标已被广泛用来估算海洋环境中土壤有机质的贡献比例[71 ~73 ] ,但有报道指出湖泊[74 ] 和海洋[75 ] 也会产生br-GDGTs.湖泊、海洋环境样品中br-GDGTs的14 C同位素特征可以为br-GDGTs土壤源指标的应用提供重要的信息.Birkholz等[48 ] 发现Lake Lusvatnet沉积物中的br-GDGTs呈现较老的14 C年龄.结合湖泊水体年轻的DOC特征[76 ] ,可明确判断该湖泊br-GDGTs来源于外源陈化土壤的输入. ...
Fluxes and distribution of tetraether lipids in an equatorial African lake: Constraints on the application of the TEX86 palaeothermometer and BIT index in lacustrine settings
1
2009
... Birhkolz等[48 ] 改进了已有的Prep-HPLC技术,成功分离富集出湖泊沉积物中br-GDGTs化合物单体.首先利用反相-HPLC(Agilent Eclipse Plus C18 column,4.6 mm×150 mm, 3.5 μm)将br-GDGTs和共流出组分分离;为了除去反相-HPLC柱分离过程中引入的共流出组分和进一步纯化,使用正相-HPLC柱(Alltech Prevail Cyano column, 2.1 mm×150 mm, 3 μm)消除可能来自流动相的外源碳(12 μg C/60 min).改进后的HPLC技术贡献的外源碳为 (0.6±0.2) μg C(其F14 C为0.5±0.2),与Hou等[14 ] 结果基本一致,其外源碳的贡献约为2 μg C(Fm值为0.30).基于br-GDGTs的BIT指标已被广泛用来估算海洋环境中土壤有机质的贡献比例[71 ~73 ] ,但有报道指出湖泊[74 ] 和海洋[75 ] 也会产生br-GDGTs.湖泊、海洋环境样品中br-GDGTs的14 C同位素特征可以为br-GDGTs土壤源指标的应用提供重要的信息.Birkholz等[48 ] 发现Lake Lusvatnet沉积物中的br-GDGTs呈现较老的14 C年龄.结合湖泊水体年轻的DOC特征[76 ] ,可明确判断该湖泊br-GDGTs来源于外源陈化土壤的输入. ...
Constraints on the sources of branched tetraether membrane lipids in distal marine sediments
1
2014
... Birhkolz等[48 ] 改进了已有的Prep-HPLC技术,成功分离富集出湖泊沉积物中br-GDGTs化合物单体.首先利用反相-HPLC(Agilent Eclipse Plus C18 column,4.6 mm×150 mm, 3.5 μm)将br-GDGTs和共流出组分分离;为了除去反相-HPLC柱分离过程中引入的共流出组分和进一步纯化,使用正相-HPLC柱(Alltech Prevail Cyano column, 2.1 mm×150 mm, 3 μm)消除可能来自流动相的外源碳(12 μg C/60 min).改进后的HPLC技术贡献的外源碳为 (0.6±0.2) μg C(其F14 C为0.5±0.2),与Hou等[14 ] 结果基本一致,其外源碳的贡献约为2 μg C(Fm值为0.30).基于br-GDGTs的BIT指标已被广泛用来估算海洋环境中土壤有机质的贡献比例[71 ~73 ] ,但有报道指出湖泊[74 ] 和海洋[75 ] 也会产生br-GDGTs.湖泊、海洋环境样品中br-GDGTs的14 C同位素特征可以为br-GDGTs土壤源指标的应用提供重要的信息.Birkholz等[48 ] 发现Lake Lusvatnet沉积物中的br-GDGTs呈现较老的14 C年龄.结合湖泊水体年轻的DOC特征[76 ] ,可明确判断该湖泊br-GDGTs来源于外源陈化土壤的输入. ...
Radiocarbon and stable-isotope geochemistry of organic and inorganic carbon in Lake Superior
1
2012
... Birhkolz等[48 ] 改进了已有的Prep-HPLC技术,成功分离富集出湖泊沉积物中br-GDGTs化合物单体.首先利用反相-HPLC(Agilent Eclipse Plus C18 column,4.6 mm×150 mm, 3.5 μm)将br-GDGTs和共流出组分分离;为了除去反相-HPLC柱分离过程中引入的共流出组分和进一步纯化,使用正相-HPLC柱(Alltech Prevail Cyano column, 2.1 mm×150 mm, 3 μm)消除可能来自流动相的外源碳(12 μg C/60 min).改进后的HPLC技术贡献的外源碳为 (0.6±0.2) μg C(其F14 C为0.5±0.2),与Hou等[14 ] 结果基本一致,其外源碳的贡献约为2 μg C(Fm值为0.30).基于br-GDGTs的BIT指标已被广泛用来估算海洋环境中土壤有机质的贡献比例[71 ~73 ] ,但有报道指出湖泊[74 ] 和海洋[75 ] 也会产生br-GDGTs.湖泊、海洋环境样品中br-GDGTs的14 C同位素特征可以为br-GDGTs土壤源指标的应用提供重要的信息.Birkholz等[48 ] 发现Lake Lusvatnet沉积物中的br-GDGTs呈现较老的14 C年龄.结合湖泊水体年轻的DOC特征[76 ] ,可明确判断该湖泊br-GDGTs来源于外源陈化土壤的输入. ...

 , 陶舒琴
, 陶舒琴

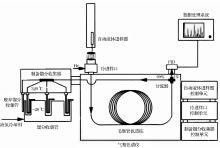
{kind=link}
{kind=link}