


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
表生环境中镁同位素的地球化学循环
[董爱国 , 朱祥坤
, 朱祥坤* ]
, 朱祥坤]
|
|


作者简介:董爱国(1982-),男,内蒙古锡林浩特人,博士后,主要从事金属同位素地球化学研究.E-mail:aiguo.dong@cags.ac.cn
近些年表生环境中镁同位素分馏取得了一系列重要研究进展,这些新认识为深入理解表生环境中镁同位素地球化学循环奠定了基础.表生环境中镁同位素的地球化学循环主要涉及风化,河流搬运,碳酸盐沉淀,水岩反应等重要地质过程.风化过程中镁同位素发生显著分馏,硅酸盐风化产物中富集重的镁同位素,轻的镁同位素易进入水体.河流搬运过程中,镁同位素不发生分馏,但外源输入可能影响水体的镁同位素组成.河水汇入海洋后,碳酸盐沉淀过程可导致轻的镁同位素以碳酸盐的形式从海水中移出.在海底高温水岩反应过程中,海水中绝大多数的镁(80%~87%)都进入岩石,循环后的热液可能富集轻的镁同位素.海底低温水岩反应过程中海水的镁可以进入岩石并形成次生矿物,此过程的镁同位素分馏主要与次生矿物的形成有关.此外,海水中的镁易与黏土矿物发生交换反应,此过程黏土矿物倾向于吸附轻的镁同位素.总之,在表生环境中上地壳的镁(26Mg约为-0.22‰)经历风化作用,河流搬运,海洋贮存,最终以碳酸盐岩(26Mg一般小于-1‰)或与玄武岩发生反应的形式重新回到岩石圈.
Mg isotope fractionation in the supergene environment have been obtained many important advances in recent years, and these new knowledge supply the clues for further understanding Mg isotope geochemical cycle in the supergene environment. Mg isotope geochemical cycle in the supergene environment involves some important geological processes, such as weathering, river transportation, carbonate sedimentation and water-rock reaction. Mg isotope fractionates dramatically in weathering process. Silicate weathering products enrich in26Mg (secondary clay minerals prefer to combine with26Mg) and24Mg prefer to be into the aqueous phase. Although there is not significant Mg isotope fractionation during river transportation, Mg isotope of river water could still be affected by the supplement of external sources. Most magnesium from river water are transported into the ocean, and then the carbonate precipitation prefers to remove24Mg from ocean as carbonate minerals. Submarine low temperature waterrock reaction could transport less magnesium from ocean into the rocks during secondary mineral formation, which is associated with Mg isotope fractionation. However, most of magnesium (80%~87%) in seawater prefer to combine with the rock in high temperature waterrock reaction and recirculated hydrothermal fluid may be enriched in24Mg. In brief, magnesium from upper crust (δ26Mg about -0.22‰) experiences weathering and transports by river water to ocean in the supergene environment. The magnesium in ocean could recycle into the rocks by the process of carbonate precipitation (δ26Mg is less than -1‰) or reacting with submarine basalt to form secondary mineral.
镁是主要的造岩元素, 也是动植物所必需的生命元素, 主要富集于地幔中, 在地壳, 水体和生物体中也广泛分布.镁可富集于岩浆过程早期结晶的矿物中(如橄榄石, 辉石等), 易在风化过程中以镁离子的形式进入水体.经水体搬运至海洋后, 可通过碳酸盐的形式沉淀或在水岩反应过程中从海水移出[1].镁在海洋中具保守性, 居留时间达1.3× 107年[2], 是表生地球化学循环过程中的主要代表性元素.
随着MC-ICP-MS测试技术的发展以及地质样品中镁同位素组成等研究工作的开展[3], 镁同位素在示踪行星演化[4], 岩浆过程[5], 风化过程[6~17], 俯冲过程[18, 19], 白云岩化过程[20~25], 矿床成因[26~28]等方面显示了重要的作用.相对于高温地球化学过程而言, 表生环境(尤其是风化和碳酸盐沉淀过程)中镁同位素分馏显著.探讨表生过程中镁同位素分馏, 不仅是应用镁同位素示踪的基础, 还对深入认识镁同位素的地球化学循环具有重要意义.本文简要总结过去十多年表生地质过程中镁同位素地球化学的研究进展, 从理论计算, 实验模拟和野外观测等方面评述风化, 河流搬运, 碳酸盐沉淀, 水岩反应等表生地质过程中镁同位素分馏, 简述表生环境中镁同位素的地球化学循环, 展望表生过程中镁同位素分馏存在的问题和镁同位素的示踪潜力.
镁有3种稳定同位素:24Mg, 25Mg和26Mg, 相对丰度分别为78.992%, 10.003%和11.005%[29].镁同位素通过δ iMg值来表示样品与标准的差异:
δ iMg (‰ ) =[(iMg/24Mg)样品/ (iMg/24Mg )标准 -1]× 1000, 其中i=25或26.
目前国际通用的镁同位素标准物质为DSM3[30].本文中报道的所有数据均以DSM3为标准.早期镁同位素数据基于国际标准物质SRM980表示, 但由于其镁同位素组成不均一, 已不适合做国际标准物质[30, 31].早期的镁同位素数据可通过以下公式转化, 其中δ 26 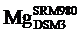
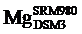
δ i 

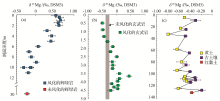
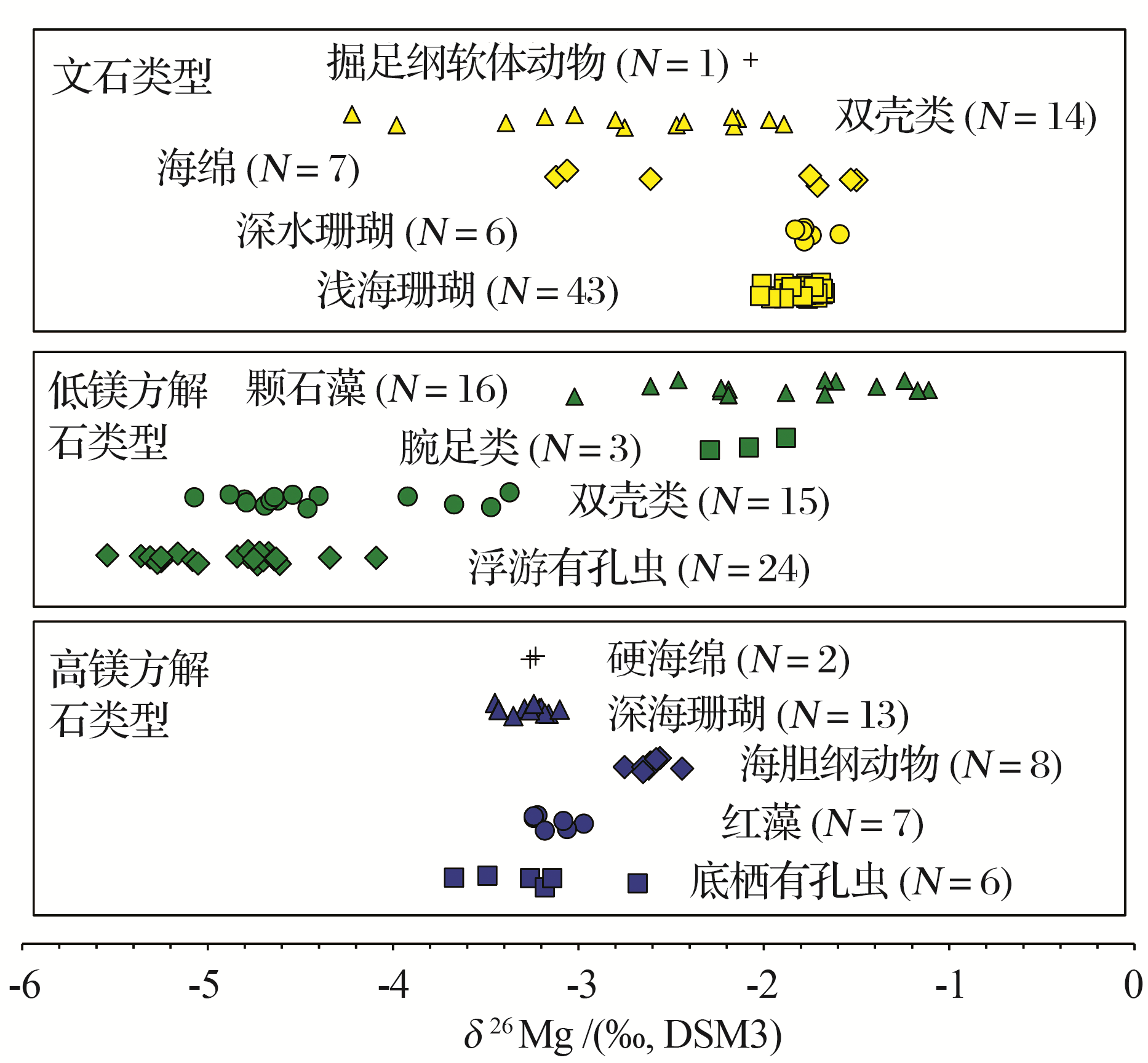
前人已对不同储库的镁同位素组成进行了较为详细的总结[33~38], 这里仅做简要概述(图1).整体硅酸盐地球具有与球粒陨石一致的镁同位素组成, δ 26Mg平均值约为-0.25‰ [41].火成岩的镁同位素组成变化范围较小, 与球粒陨石基本一致.与火成岩相比, 风化产物(黄土, 岩石风化壳等)的镁同位素组成变化较大, 总体上富集重的镁同位素[17, 71, 72].碳酸盐岩整体上富集轻的镁同位素, 其中白云岩镁同位素组成比灰岩稍重[56, 58, 60, 61, 73, 74].现代海水的镁同位素组成均一, 约为-0.82‰ [64, 65].河水的镁同位素组成变化范围较大, 与流域岩性, 风化程度等因素相关, 世界主要河流的镁同位素平均组成约为-1.09‰ ± 0.05‰ [63].生物体整体上富集轻的镁同位素且变化较大, 其δ 26Mg值可达-5.6‰ 左右[45, 48, 49].
风化过程是最基本的地质过程之一, 是连接岩石圈与水圈--大气圈--生物圈的桥梁.陆壳风化过程产生的镁离子是海水镁的主要来源.一般来说, 岩石风化过程主要受控于4种化学作用:溶解作用, 水解作用, 氧化还原作用和离子交换作用[75], 其中镁不涉及氧化还原作用.流域岩石中硅酸盐矿物主要涉及水解作用, 碳酸盐矿物主要与溶解作用有关.硅酸岩风化过程形成的次生矿物主要为黏土矿物, 其对镁离子的影响主要体现在两方面, 一方面镁离子可直接进入黏土矿物的晶格, 另一方面镁离子可被吸附在黏土矿物的表面和层间.本文主要从理论计算, 实验模拟和野外风化剖面观测等方面来讨论风化过程中镁同位素分馏.
虽然不同模型所计算的矿物间镁同位素分馏程度略有不同, 但镁同位素分馏趋势基本是一致的.不同矿物的镁同位素组成特征与矿物自身的晶体结构, 镁离子与其他离子化学键的强度密切相关.依据密度泛函微扰理论计算的矿物镁同位素组成具有以下特征[76], 即:Δ 26Mg方解石-水 < Δ 26Mg菱镁矿-水 < Δ 26Mg白云石-水 < < 0, 说明碳酸盐溶解过程中水体相对碳酸盐富集重的镁同位素.在次生矿物形成过程中, 次生矿物的类型决定了镁同位素的分馏.例如黏土是硅酸盐风化过程的主要次生矿物.虽然目前鲜有关于黏土矿物镁同位素组成的计算, 但从理论上可以推测黏土矿物晶格中的镁离子倾向于富集重的镁同位素(相对于水体).
目前风化过程中镁同位素分馏的实验研究主要分为2类, 一类为限定条件下直接观测天然岩石样品的水岩反应, 例如花岗岩或玄武岩的水岩反应.花岗岩水岩反应过程中(pH=1, T=25℃ ), 流体的镁同位素组成明显受控于不同矿物(例如绿泥石, 黑云母, 角闪石等)的溶解[77], 表明原生矿物溶解对于风化过程中镁同位素分馏具重要作用.玄武岩水岩反应实验可分为原生矿物溶解实验和次生矿物形成实验, 所选样品均为玄武质玻璃和橄榄石.在原生矿物溶解实验中体系较低的pH(2~4)确保了实验过程中不形成二次矿物; 而在二次矿物形成实验中体系较高的pH(10~11)促进了实验过程中二次矿物的形成[78].玄武质玻璃和橄榄石溶解过程中(pH为2~4), 溶液的镁同位素组成比原生矿物轻且随着时间的推移逐渐加剧, 表明轻的镁同位素优先进入流体相.在次生矿物形成实验中(pH为10~11), 随着次生矿物的形成流体镁同位素组成逐渐变重, 表明流体的镁同位素组成主要与次生矿物形成有关(纤蛇纹石形成过程优先结合轻的镁同位素).上述实验虽然证实了风化过程镁同位素分馏的与原生矿物溶解和次生矿物形成密不可分, 但其实验条件, 风化产物与天然风化过程差别较大, 说明风化过程中镁同位素分馏仍需根据实际情况来综合考虑.
另一类实验以直接观测次生矿物的镁同位素行为为主.黏土矿物是硅酸盐岩风化过程中形成的典型次生矿物, 主要为伊利石, 高岭石和蒙脱石, 其中高岭石镁含量较低(< 0.1%), 伊利石和蒙脱石含镁量相对较高(> 1%).黏土矿物的阳离子吸附量(CEC)自蒙脱石(CEC为60~100 mmol/100g)经伊利石(CEC为25~40 mmol/100g)至高岭石(CEC为3~15 mmol/100g)依次减小.一般来说, 黏土矿物对于镁离子的吸收存在两种形式, 一部分镁离子直接进入黏土矿物的八面体晶格中, 另一部分镁离子被吸附在黏土矿物的表面或层间.黏土矿物的醋酸淋洗实验表明, 残余黏土矿物(晶格中的镁离子)富集重的镁同位素而淋洗液中富集轻的镁同位素.此外, 黏土矿物与中性氯化铵溶液的离子交换实验[79]表明, 交换后溶液的镁同位素组成显著轻于残留黏土矿物的镁同位素组成, 说明以吸附形式存在的镁离子富集轻的镁同位素.黏土矿物所表现出的镁同位素组成是上述两种镁同位素特征(晶格中的镁和吸附的镁)的叠加, 这也可能是自然界中黏土矿物(高岭石, 伊利石, 蒙脱石)的镁同位素组成具有较大变化范围(可达2‰ [79])的原因.上述实验对认识风化过程中镁同位素分馏具有重要意义, 也为我们深入理解不同风化剖面镁同位素组成的变化提供了理论依据.
风化剖面是观测风化作用最直接的场所.图2为不同类型风化壳的镁同位素组成.辉绿岩风化壳中风化产物的镁同位素组成明显重于原岩, 且随着风化程度的增强, 不同层位风化产物的镁同位素组成逐渐变重(δ 26Mg值从-0.22‰ 逐渐增加到0.65‰ [11]).此辉绿岩风化壳的镁同位素组成与次生矿物的形成密切相关, 即次生矿物富集重的镁同位素而相应的水体富集轻的镁同位素.在火山沉积风化层序中也观测到含镁次生矿物的镁同位素组成比源岩安山岩重[10].再者, 强烈的风化作用能够促进风化残余相的镁同位素组成发生显著变化, 这一现象在矾土风化剖面(源岩为玄武岩)上有明显记录.矾土风化剖面的残留风化产物相对于源岩具有更重的镁同位素组成, 并在某些层位观测到目前风化残留产物中最重的镁同位素组成(δ 26Mg值可达1.8‰ [8]).此外, 矾土风化剖面的残留风化产物镁同位素组成与次生矿物(三水铝石)含量也有一定相关性(相关系数R2约为0.55):风化残留产物中三水铝石越高镁同位素组成越重.虽然辉绿岩风化壳和矾土风化剖面的风化程度差异明显, 但风化过程所引起的镁同位素分馏程度却相差不大, 都介于0.4‰ 以内[8, 11].与矾土风化剖面类似, 我国海南玄武岩风化剖面也经历了强烈的风化作用.风化产物的氧化镁含量不足1%(源岩约为7%), 且氧化镁含量随剖面深度的增加逐渐增高(0~3 m趋势明显, 3~4.5 m趋势不明显; 见图2).关于这一现象, 早期的研究[9]认为次生矿物(例如高岭土)表面吸附重的镁同位素, 风化过程中由于表面重同位素被淋洗掉, 导致残留产物镁同位素组成显著变轻.但后续黏土矿物镁同位素分馏的模拟实验[79]给出了不同的解释, 即海南玄武岩风化剖面中残留产物(高岭土)镁同位素组成由吸附的镁离子含量和矿物晶格内镁离子含量的比例所决定(吸附的镁离子具相对轻的镁同位素组成, 但矿物晶格内镁离子具相对重的镁同位素组成).当吸附的镁离子为主导时, 风化残余则表现出轻的镁同位素组成.从目前的研究来看, 基于黏土矿物分馏实验的解释更合理.
在页岩风化剖面中, 风化后土壤比于孔隙水富集重的镁同位素, 说明了硅酸盐风化过程中轻的镁同位素优先进入水体.然而页岩风化剖面的镁同位素组成与风化程度呈反相关:即随着风化程度的增强, 不同层位风化产物的镁同位素组成逐渐变轻(δ 26Mg值从0.35‰ 逐渐降低到0.22‰ [68].此趋势产生的原因可能是形成了具有较轻镁同位素组成的蛭石(次生矿物, 与黏土矿物类似)或部分重的镁同位素富集于细颗粒并在风化过程中以悬浮体的形式进入水体导致风化产物相对富集轻的镁同位素.
总之, 风化产物的镁含量较高时, 风化过程中形成的黏土矿物主要以富集重的镁同位素组成为特征; 风化产物的镁含量很低时, (如上述矾土, 海南玄武岩风化剖面), 形成的黏土矿物镁同位素组成则由吸附的镁(轻的镁同位素)和矿物晶格内镁(重的镁同位素)的比例决定, 且此类风化产物的镁同位素组成更易受到外部物质来源(如风成沉积物)等因素的影响.
此外, 黄土剖面的镁同位素组成与古风化作用存在耦合关系[79](图2).黄土层中镁同位素组成略轻于古土壤层, 且两者均轻于上地壳的镁同位素组成.相对于干冷的冰期, 间冰期的风化作用较强, 易导致黄土发生成土作用形成古土壤.相对于古土壤层, 黄土层具有较多的碳酸盐矿物(具有较高的Ca/Ti比值)且更富集轻的镁同位素.在遭受较强风化作用时黄土中碳酸盐矿物优先溶解并形成具较重镁同位素组成的古土壤.因此, 洛川黄土剖面的镁同位素组成特征表明风化过程中原生碳酸盐矿物的溶解是控制黄土剖面中镁同位素组成变化的主要因素且镁同位素组成的变化与风化程度密切相关.
综上所述, 原生矿物溶解和次生矿物形成是控制风化过程中镁同位素分馏的主要因素.由于源岩类型的不一致, 风化作用程度的不同, 风化过程中产生的次生矿物类型差异较大.对于次生矿物的镁同位素行为的实验模拟将有助于深入理解风化过程中镁同位素的地球化学行为.
 | 图2 不同风化剖面中镁同位素组成 (a)为辉绿岩风化剖面, 数据来源于文献[11]; (b)为过度风化的玄武岩剖面, 数据来源于文献[9]; (c)为洛川黄土剖面, 数据来源于文献[79]Fig 2 Mg isotopic composition in weathering profiles. (a)Represents the diabase weathering profile, data source from reference[11]; (b) represents the intense weathering profile of the basalt, data source from reference[9]; (c)represents Luochuan loess profile, data source from reference[79] |
陆壳经过风化后大量的镁离子进入河水, 之后经河流输送至海洋.河流输送的镁是海水镁的主要来源.虽然在河流搬运过程中镁同位素不发生显著的分馏, 但外源的补给仍可能影响河水的镁同位素组成.目前的研究表明[63], 河水的平均镁同位素组成(δ 26Mg约为-1.09‰ )比海水(δ 26Mg为-0.82‰ )要轻.
源岩是风化过程的物质基础, 其岩石类型对于风化后水体的镁同位素组成具重要影响.一般来说, 对于硅酸岩流域水体的镁同位素组成比源岩硅酸岩轻, 而碳酸盐流域水体的镁同位素比源岩碳酸盐岩略重, 但是碳酸盐岩流域水体的镁同位素组成仍比硅酸岩流域水体要轻[63].由于大型河流水体的镁同位素组成变化较小且入海水量巨大, 故可近似认为世界河水整体上具有较轻的镁同位素组成, 其平均δ 26Mg值约为-1.09‰ [63].此外, 格陵兰岛冰川河水与底质沉积物之间的镁同位素分馏(Δ 26Mg冰川河水-底质沉积物 = -0.7‰ )显著大于非冰川河水与其底质沉积物之间的镁同位素分馏(Δ 26Mg非冰川河水-底质沉积物 = -0.2‰ ), 其中底质沉积物的镁同位素组成均为-0.4‰ , 冰川河水的镁同位素组成(δ 26Mg为-1‰ ~ -1.3‰ )比非冰川河水轻(δ 26Mg约为-0.6‰ ).这一现象可能与风化过程中冰川河水流域源岩的碳酸盐矿物(具有较低的镁同位素组成)优先溶解有关[7].
在河流搬运过程中, 某些河水的镁同位素组成可能受外源补给(风成沉积, 雨水, 海水, 植被等)的影响.风成沉积的供给对某些镁含量较低的流域水体也有重要的影响.一般风成沉积物中硅酸盐的镁同位素组成与上地壳接近(-0.22‰ )[80], 而碳酸盐的镁同位素组成显著轻于上地壳.风成沉积供给的镁能够直接在河流中与水体作用, 影响水体的镁同位素组成, 但具体的情况应考虑风成沉积的主要矿物类型[10].如果风成沉积的镁主要以碳酸盐的形式存在, 那么进入河水的碳酸盐易发生溶解进而影响水体的镁同位素组成.除了风尘沉积供给之外, 雨水和海水也可能影响河水的镁同位素组成.某些热带流域(例如波多黎各)降水量较大同时化学风化较为强烈, 这种情况下雨水供给对流域河水的镁同位素组成有重要影响[81].不仅是热带河流, 冰川河水镁同位素组成也易受到降水的影响[10].这一特征可能与冰川河流具有较低的镁含量有关.另外, 法属瓜德罗普岛的火山岩土壤剖面所遭受的化学风化作用强烈且土壤中镁元素含量严重亏损.土壤中易迁移镁元素的镁同位素组成介于海水和源岩之间.如果假设易迁移的镁均来源于海水, 那么以此估算的土壤镁同位素组成与实际测定的镁同位素组成在测试误差内一致, 说明了海洋飞沫可能是土壤中可迁移镁的主要来源[72].目前河流流域镁同位素研究表明, 虽然植物对于大型流域水体中镁同位素组成的影响较小, 但对于局部地区水体或土壤的镁同位素组成可产生重要影响.整体而言, 植物体以富集轻的镁同位素为特征[29, 69, 70, 82].植被覆盖的小型流域中具较轻镁同位素组成的枝叶会以落叶的形式重新回到土壤中.例如春季湿润时期, 土壤孔隙水的镁同位素组成与雨水或者落叶接近[83], 表明小型流域内水体的镁同位素组成易受植物或雨水的影响.但是在大型流域中, 植物对河水镁同位素组成的影响很小.
镁同位素地球化学循环过程中, 碳酸盐的沉淀是海水镁进入沉积岩的主要方式之一.此过程碳酸盐优先结合水体中较轻的镁同位素, 可能是导致海水的镁同位素组成(-0.82‰ )略重于河水(-1.09‰ )的主要原因.碳酸盐沉淀过程中镁同位素分馏可分为无机沉淀和有生物体参与的沉淀过程.白云石, 方解石, 菱镁矿, 文石将在无机沉淀过程中讨论, 这些碳酸盐岩具有明显轻于海水的镁同位素组成.其中文石和水菱镁矿沉淀过程中镁同位素分馏程度最小, 方解石沉淀过程中镁同位素分馏程度最大[74].碳酸盐沉淀过程可能受沉淀矿物类型, 温度, 沉淀速度, 非晶质碳酸盐等因素影响.生物成因碳酸盐形成过程受无机沉淀和生物过程的双重影响.生物过程能导致较大的镁同位素分馏, 例如有孔虫具相对很轻的镁同位素组成, 其δ 26Mg值可达-5.6‰ 左右, 明显小于沉积的碳酸盐[74].整体而言, 生物成因碳酸盐比无机碳酸盐具有更复杂的镁同位素组成.
本文讨论的无机碳酸盐主要包括方解石, 文石, 菱镁矿, 白云石, 水合菱镁矿等, 其理论计算和模拟实验的镁同位素分馏结果见图3.目前方解石沉淀模拟实验表明, 不同实验条件(主要为2类, 一类是模拟过程中化学参数基本保持不变, 另一类是设定初始条件后自然反应)所测定的镁同位素在溶液和方解石(CaCO3)中的分馏程度(Δ 26Mg方解石-溶液)存在差异:Immenhauser[61]等开展的模拟实验发现Δ 26Mg方解石-溶液 介于-2.3‰ ~-1.6‰ , 考虑到沉淀速率对Δ 26Mg方解石-溶液的影响, 结合野外观测特征推荐常温下Δ 26Mg方解石-溶液约为-2‰ ; Saulnier等[85]开展的模拟实验结果表明Δ 26Mg方解石-溶液 为-2.5‰ ~-1.3‰ 且与方解石生长速率有关, 结合前人研究结果推荐25℃ 下Δ 26Mg方解石-溶液约为-2.1‰ ; Mavromatis等[86]开展的模拟实验进一步表明在不同的沉淀速率下(介于10-8.3~10-6.6 mol/(m2 S)之间)Δ 26Mg方解石-溶液 介于-3.2‰ ~-1.9‰ , 沉淀速度与分馏程度(|Δ 26Mg方解石-溶液|)正相关:Δ 26Mg方解石-溶液 = 0.7918 (± 0.0452) × Log rp + 3.2366 (± 0.336) (rp为沉淀速率, 介于10-8.3~10-6.6 mol/(m2 S)之间).其认为大部分沉积碳酸盐或者生物成因碳酸盐的镁同位素组成都受到动力分馏过程的影响, 若沉积速率足够低(小于10-8.5 mol/(m2 S)), Δ 26Mg方解石-溶液的平衡分馏值可达-3.5‰ .上述方解石沉淀的模拟实验结果说明了:① 方解石沉淀过程中存在动力学分馏且动力学分馏过程中方解石相对富集重的镁同位素组成; ② Δ 26Mg方解石-溶液值至少可达-2‰ , 若沉积速率足够小Δ 26Mg方解石-溶液的平衡分馏值可达-3.5‰ .与上述方解石沉淀实验(沉淀速率控制了镁同位素的分馏)不同, Li等[87]开展的方解石沉淀模拟实验(设定初始条件后自然反应)表明在不同的温度下(4~45℃ )Δ 26Mg方解石-溶液在-2.7‰ ~-2.22‰ 之间变化且其变化程度(|Δ 26Mg方解石-溶液|)随温度的升高而降低.方解石沉淀分馏程度与温度的关系为0.011‰ /℃ , 与早期野外观测所得的结果(沉淀分馏程度与温度的关系为0.04‰ /℃ [56])趋势相一致.目前关于方解石与水溶液达到平衡时的分馏程度存在不同的认识, 即模拟实验所推算的Δ 26Mg方解石-溶液平衡分馏值介于-3.5‰ ~ -2‰ , 远小于理论计算的结果[76, 84, 88].野外观测的结果似乎更支持模拟实验所估算的Δ 26Mg方解石-溶液平衡分馏值.总体而言, 自然界中大多数方解石的沉淀过程可能并未达到平衡.
文石(CaCO3)与方解石化学成分一致, 但结构却不同, 也是一种重要的沉积碳酸盐矿物.目前文石沉淀过程的模拟实验仅有一例, 模拟实验在常压, 22~55 ℃ , 与海水性质类似的水体(Mg/Ca = 5 或者10)中进行, 属于"设定初始条件后自然反应"类型的实验[89].实验结果表明, Δ 26Mg文石-溶液为-1.09‰ ~-0.81‰ 且与温度密切相关:Δ 26Mg文石-溶液 = 1.67 (± 0.36) - 0.82 (± 0.11) × 1000/T (T为温度, 单位为开尔文), 沉淀分馏程度与温度的关系为0.008‰ /℃ ~0.01‰ /℃ .而理论计算25 ℃ 平衡时Δ 26Mg文石-溶液值约为-9.8‰ , 与实验模拟和野外观测所得到的Δ 26Mg文石-溶液差异巨大.从目前的研究来看, 理论计算所得的Δ 26Mg文石-溶液值可能存在较大的问题, 可能与镁离子在文石(理想的文石和自然界存在的文石)中占位的差异有关.
白云石(MgCa(CO3)2)是最重要的富镁碳酸盐矿物, 广泛分布于古老的碳酸盐岩地层中.白云石在常温常压下难于直接沉淀形成, 而易产生于热液交代或二次白云岩化过程中.Li等在热液条件下开展了白云石形成模拟实验[90], 实验温度分别为130, 160和220 ℃ , 初始矿物也略有不同(分别有文石, 方解石, 三水菱镁矿等).扫描电镜和X衍射分析显示绝大部分实验中均产生了白云石(仅有一个实验形成了无序的白云石).实验结果表明, 白云石形成时镁同位素的分馏基本达到平衡, 在130 ℃ 时, Δ 26Mg白云石-溶液为-0.93‰ ; 在160 ℃ 时, Δ 26Mg白云石-溶液为-0.84‰ ; 在220 ℃ 时, Δ 26Mg白云石-溶液为-0.65‰ .总体而言, 分馏程度与温度呈现较好的相关性:Δ 26Mg白云石-溶液 = -0.1554 (± 0.0096) × 106/T2 (T为温度, 单位为开尔文).实验估算的Δ 26Mg白云石-溶液值介于理论计算范围之内[76, 87], 且25℃ 时Δ 26Mg白云石-溶液约为-1.75‰ .此值略小于深海沉积物中经白云石和孔隙水计算的Δ 26Mg白云石-溶液[25](Δ 26Mg白云石-溶液为-2.0‰ ~ -2.7‰ ), 也与理论计算的结果[76, 87, 88]差异较大.
 | 图3 碳酸盐沉淀过程中镁同位素分馏的理论估算和实验测定(修改自参考文献[88])Fig 3 Theoretical estimation and experimental determination of Mg isotope fractionation in carbonate precipitation (modified from reference[88]) |
菱镁矿(MgCO3)是重要的镁碳酸盐, 其在常温常压下难于形成.因此, Pearce等[91]在120~200 ℃ , CO2压力为15~30个大气压条件下开展了菱镁矿的合成实验并测定了其镁同位素组成.在150 ℃ 时, Δ 26Mg菱镁矿-溶液为-1.19‰ ; 在200 ℃ 时, Δ 26Mg菱镁矿-溶液为-0.88‰ , 暗示了菱镁矿的分馏程度与温度具有一定关系.依此推算, 25℃ 时Δ 26Mg菱镁矿-溶液约为-2.3‰ .镁同位素达到平衡时, 理论计算和实验模拟所得的Δ 26Mg菱镁矿-溶液值较为接近, 指示了平衡分馏时Δ 26Mg菱镁矿-溶液约为-2‰ (25 ℃ ).
水菱镁矿(Mg5(CO3)4(OH)2· 4H2O)在现代盐湖的叠层石中(例如土耳其萨尔达盐湖等)广泛分布, 是现代沉积环境中富镁碳酸盐的重要组成, 在一定的条件下脱水可形成菱镁矿.Shirokova等[73]开展了水菱镁矿的沉淀实验, 并在部分实验中加入了蓝细菌, 模拟实验沉淀的矿物主要为水菱镁矿和球碳镁石(Mg5(CO3)4(OH)2· 5H2O).实验结果表明, Δ 26Mg水菱镁矿-溶液值为-1.1‰ ~-0.9‰ , 与其在野外观测(土耳其萨尔达盐湖)的结果一致.同时蓝细菌的加入促进了水菱镁矿的形成, 但并未改变其同位素组成, 这一结果与模拟实验结果[92]一致, 说明平衡时Δ 26Mg水菱镁矿-溶液约为-1‰ (常温下).
总之, 碳酸盐矿物相对于沉淀水体整体富集轻的镁同位素组成.从目前的模拟实验可推知:25 ℃ 时Δ 26Mg方解石-溶液至少小于-2‰ , 甚至于可达到-3.5‰ ; 25 ℃ 时Δ 26Mg菱镁矿-溶液约为-2.3‰ ; 25 ℃ 时Δ 26Mg白云石-溶液约为-1.75‰ ; 常温时Δ 26Mg水菱镁矿-溶液约为-1‰ ; 25 ℃ 时Δ 26Mg文石-溶液约为-1‰ .其中平衡时理论计算, 实验模拟和野外观测的Δ 26Mg方解石-溶液, Δ 26Mg文石-溶液, Δ 26Mg白云石-溶液仍存在较大的差异.这种差异产生的原因一方面可能为上述碳酸盐沉淀过程中至少有一个过程(模拟或实际观测)的镁同位素分馏未能达到平衡, 另一方面可能实际碳酸盐样品与理论计算, 模拟实验的理想碳酸盐沉淀过程略有不同.碳酸盐沉淀过程中镁同位素的分馏不仅取决于所形成碳酸盐的晶体结构, 而且还与水合镁离子(水中的镁离子易与水结合形成水合镁离子)在不同晶体表面脱水过程有关.
总体而言, 沉淀过程中轻的镁同位素趋向于在碳酸盐中富集.野外观测, 实验模拟和理论计算的碳酸盐岩镁同位组成表明, 不同的碳酸盐岩矿物具有显著的镁同位素组成差异, 说明矿物类型是控制不同碳酸盐岩镁同位素组成的首要因素[53].结合目前的实验模拟, 野外观测以及理论计算结果可推断碳酸盐矿物与溶液的分馏程度(|Δ 26Mg碳酸盐岩矿物-溶液|)如下:|Δ 26Mg文石-溶液| ≈ |Δ 26Mg水合菱镁矿-溶液| < |Δ 26Mg白云石-溶液| ≈ |Δ 26Mg菱镁矿-溶液| < |Δ 26Mg方解石-溶液|.
碳酸盐沉淀过程中镁同位素分馏程度与溶液的Mg/Ca比值, 溶液饱和度, 溶液pH无关, 但明显受到温度和沉淀速率的影响[74, 85~87, 91, 93].早期对钟乳石的野外观测表明, 钟乳石的镁同位素分馏与温度有关且分馏程度约为0.04‰ /℃ [56].但是, 此过程中Δ 26Mg方解石-溶液的变化程度(4~17 ℃ , Δ 26Mg方解石-溶液为-2.8‰ ~-2.57‰ )与测试精度非常接近.其他实验模拟的结果显示Δ 26Mg碳酸盐-溶液与温度正相关, 方解石随温度变化的分馏程度约为0.011‰ /℃ [87], 而文石随温度变化的分馏程度约为0.009‰ /℃ [89].白云石的分馏程度也与温度相关, 其随温度变化的分馏程度约为0.011‰ /℃ [90].在菱镁矿沉淀实验中, 如果根据实验结果进行外推的话, 镁同位素组成也与温度有关联, 分馏程度约为0.014‰ /℃ [91].从理论上来说, 镁同位素分馏是温度的函数, 随着温度的升高分馏程度逐渐降低[94], 但是上述实验中观测到的镁同位素分馏程度与温度的趋势远小于理论计算值.其可能产生的原因为:① 理论计算所用的矿物结构为标准矿物结构, 与实际碳酸盐岩有差异.② 上述碳酸盐模拟实验过程中可能受到沉淀速率的影响直接结合水体中水合镁离子或碳酸盐沉淀过程未达到平衡[85].此外, 即使考虑温度对碳酸盐沉淀的影响, 温度波动10 ℃ 可能引起镁同位素组成的变化约为0.1‰ .这样的话, 温度的变化可能并不是导致地质历史时期同类型碳酸盐岩样品镁同位素组成具较大变化特征(图1)的原因.
沉淀速率能够引起碳酸盐沉淀过程中镁同位素分馏, 在部分模拟实验中[42, 85, 86]被观测到.模拟实验中方解石的镁同位素组成虽与温度, Ca/Mg, pH等无关, 但是沉淀速率限制了镁同位素的分馏, 较小的Δ 26Mg方解石-溶液分馏可能由于沉淀过程并未达到平衡[86].这一推论也得到野外观测的支持, 在洞穴钟乳石中镁同位素组成随着沉淀速率的增加而变重[56].一般来说, 动力分馏过程中轻同位素更易迁移, 即形成的碳酸盐应具有更轻的镁同位素组成.实际观测结果却相反, 沉淀速率的增加导致了镁同位素变重.这种现象产生的主要原因可能与镁元素在水中具有较高的吉布斯自由能, 更倾向于和水结合形成水合镁离子有关.碳酸盐表层交换模型[85, 95]推测, 溶液中的镁离子进入到碳酸盐矿物中可能存在2个过程.过程① :镁离子在水体形成水合离子, 此过程镁同位素不发生分馏.过程② :之后水合镁离子被碳酸盐矿物表面所吸附, 并在矿物表面形成类似薄膜的扩散带, 轻的镁同位素向矿物内部扩散, 此过程中发生显著的镁同位素分馏.在沉淀速率较快时, 过程② 中镁离子未达到扩散平衡, 导致沉淀的碳酸盐中镁同位素组成变重.另外, 由于目前观测到不同的碳酸盐矿物具有显著不同的镁同位素组成, 那么碳酸盐岩形成过程中是否能直接形成标准的碳酸盐晶体也可能对镁同位素分馏有很大的影响[53].例如方解石在沉淀过程中可能先形成一些非晶质的碳酸盐物质, 之后转变为方解石, 而由于晶体结构的差异, Δ 26Mg非晶质碳酸盐-溶液与Δ 26Mg方解石-溶液可能相差较大.在地质历史时期, 方解石沉淀过程中先形成的非晶质高钙碳酸盐(后期完全转化为方解石)可能会影响最终方解石的镁同位素组成.
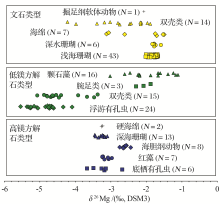
生物成因碳酸盐的镁同位素组成见图4, 相对于无机碳酸盐而言, 生物碳酸盐具有更复杂的镁同位素组成特征.随着生物种类的不同其镁同位素组成差异明显, 总体而言方解石型生物成因碳酸盐比文石型生物成因碳酸盐具有更轻的镁同位素组成.这种复杂的镁同位素组成特征可能由两方面原因引起:① 碳酸盐沉淀过程中镁同位素的分馏程度与矿物类型, 温度, 沉淀速率等密切相关, 还可能沉淀非晶质的碳酸盐矿物, 进而影响生物碳酸盐的镁同位素组成.② 部分生物体能够吸收镁离子, 这种吸收过程中发生镁同位素分馏, 优先吸收轻的镁同位素.同时生物吸收镁离子的过程还可能因生物种属的差异而发生变化.
生物过程中镁同位素的分馏是控制生物成因碳酸盐中镁同位素组成的主要因素.例如浮游有孔虫的镁同位素组成比珊瑚更轻且变化范围更大(图4).这可能是由海洋生物对镁的利用方式不同所引起的.对海洋生物体而言, 至少存在着两种镁同位素利用机制[52], 一种是将镁吸收利用(优先吸收轻的镁同位素, 如部分有孔虫等), 易产生较大的镁同位素分馏(依赖于生物种属); 另一种是将镁"吸附"在生物体周围(例如部分珊瑚), 其作用与碳酸盐岩沉淀过程相类似, 产生的镁同位素分馏与碳酸盐岩沉淀过程中镁同位素分馏相近.
矿物类型也是控制生物碳酸盐中镁同位素组成的主要因素.矿物类型主要可以分为两种:类方解石型(低镁生物成因碳酸盐, 高镁生物成因碳酸盐)和类文石型(文石型生物成因碳酸盐).无机碳酸盐沉淀实验, 理论计算以及野外观察表明, |Δ 26Mg文石-溶液| < < |Δ 26Mg方解石-溶液|, 也就是说低镁生物碳酸盐和高镁生物碳酸盐的镁同位素组成显著轻于文石型生物成因碳酸盐.例如:浮游有孔虫(低镁生物成因碳酸盐), 底栖有孔虫(高镁生物成因碳酸盐)的镁同位素组成显著轻于浅水珊瑚(文石型生物成因碳酸盐).
沉淀非晶质的碳酸盐也可能是影响生物碳酸盐镁同位素分馏的重要因素.非晶质的碳酸盐沉淀可能会优先吸收重的镁同位素, 导致相应的生物碳酸盐中镁同位素组成变重, 例如海胆类生物碳酸盐的镁同位素组成可能与沉淀非晶质碳酸盐有关[46, 51].非晶质碳酸盐的沉淀过程还需更多后续的实验工作来支撑.海水的温度, 盐度以及碳酸盐沉淀速率等因素都可能影响生物碳酸盐岩的镁同位素组成, 且随着生物种类的不同而略有差异.总之, 生物碳酸盐的镁同位素组成是由多种因素控制的, 对其镁同位素组成的探讨不仅需要结合无机碳酸盐沉淀过程, 还需考虑生物的种属以及相应的生物过程.
海水直接或下渗与海底玄武岩反应, 这一过程在全球海底广泛发育并产生显著的物质和能量交换, 进而影响整个海洋的化学组成[96].水岩反应可以分为高温和低温两个过程.高温水岩反应过程中, 海水下渗与岩石发生反应, 导致绝大多数的镁(80%~87%)从海水中移出[96].由于高温热液含镁量很低且可能富集轻的镁同位素(仅有一个数据[97]), 那么这个过程中可能存在镁同位素分馏, 但其对于海水镁同位素组成的影响(约为河流输入贡献的20%)可能较小.
低温水岩反应过程中海水的镁仅有部分进入岩石并生成次生矿物, 此过程镁同位素可能发生分馏, 其分馏过程主要受次生矿物的形成所控制.目前研究并未对低温水岩反应过程中镁同位素分馏达成共识.在太平洋东部IODP1526孔中蚀变的大洋玄武岩和辉长岩的镁同位素组成相对均一(-0.25‰ ± 0.11‰ ), 具有与地幔橄榄岩相一致的镁同位素组成[98].这一特征表明海底低温蚀变过程不能显著改变玄武岩和辉长岩(全岩尺度上)的镁同位素组成.其他学者[99]认为在岩石蚀变过程中蛇纹石化作用不发生镁同位素的分馏, 但在后期碳酸盐化作用下内部矿物之间(菱镁矿, 滑石等)发生显著的镁同位素分馏.
海洋中镁主要来源为河流的输入, 同时地下水的输入也是海水镁的来源.河水的镁同位素具有较大的范围, 整体上具有较轻的镁同位素组成, 其平均值(δ 26Mg值)约为-1.09‰ [63].地下水的输入量不到河流输入的10%, 其镁同位素组成可能比海水轻[63].
海洋中镁的主要归宿分别为碳酸盐岩, 海底岩石对镁的吸收(水岩反应过程), 与黏土发生离子交换等.碳酸盐沉淀过程是镁从海水迁移进入岩石圈的重要过程, 此过程中发生显著的镁同位素分馏且碳酸盐比海水更富集轻的镁同位素.由于方解石含镁量很低, 菱镁矿等沉淀的总量很小, 故一般可以近似地用白云石来代表碳酸盐沉淀过程.目前的研究表明白云石平均的镁同位素组成约为-1.76‰ [60].
水岩反应过程也是水圈与岩石圈物质交换的重要过程.高温水岩反应中参与海底热液循环的海水直接与岩石反应, 其中绝大多数(80%~87%)的镁直接被岩石吸收.若未完全吸收, 则镁同位素发生分馏且重的镁同位素可能优先进入岩石中, 轻的镁同位素以热液的形式返回至海水.低温玄武岩蚀变过程元素交换过程存在着较多的争议.目前通过大洋钻探研究发现, 总体来看低温玄武岩蚀变过程中洋壳向海水释放镁离子, 但含量较低(不到河流输入的2%[100]).即使洋壳蚀变过程中可能存在着较大的镁同位素分馏, 但其对于海洋镁同位素组成的影响很小.海水与黏土矿物的离子交换过程可能存在着分馏(轻的镁同位素倾向于进入黏土矿物层间并被吸附)或分馏很小[78].同样由于海水与黏土矿物离子交换过程迁移的镁总量较小[100], 其对于海水的镁同位素组成影响也很小.
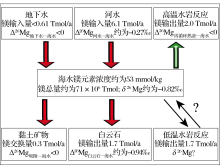
现代海水具有稳定的同位素组成-0.82‰ , 河水的平均镁同位素组成约为-1.09‰ .两者镁同位素差异说明海洋中镁同位素的输入与输出的量不平衡, 基于箱式模型(图5)可简单表示为:
T输入 = J河水 × |Δ 26Mg河水-海水| + J地下水 × |Δ 26Mg地下水-海水|
T输出 = J白云石 × |Δ 26Mg白云石-海水| + J其他 × |Δ 26Mg其他-海水| - J高温热液 × |Δ 26Mg高温热液-海水|× K热液
其中, J代表海水中镁的输入量, 例如J河水代表河流的镁输入通量; T输入代表输入的镁同位素变化量; T输出代表输出的镁同位素变化总量; Δ 26Mg物质-海水 代表某物质与海水镁同位素组成的差异, 例如Δ 26Mg白云石-海水 代表白云岩与海水镁同位素组成的差异; K热液代表与海底岩石反应后再次进入海水的热液所占比例, 取0.1.
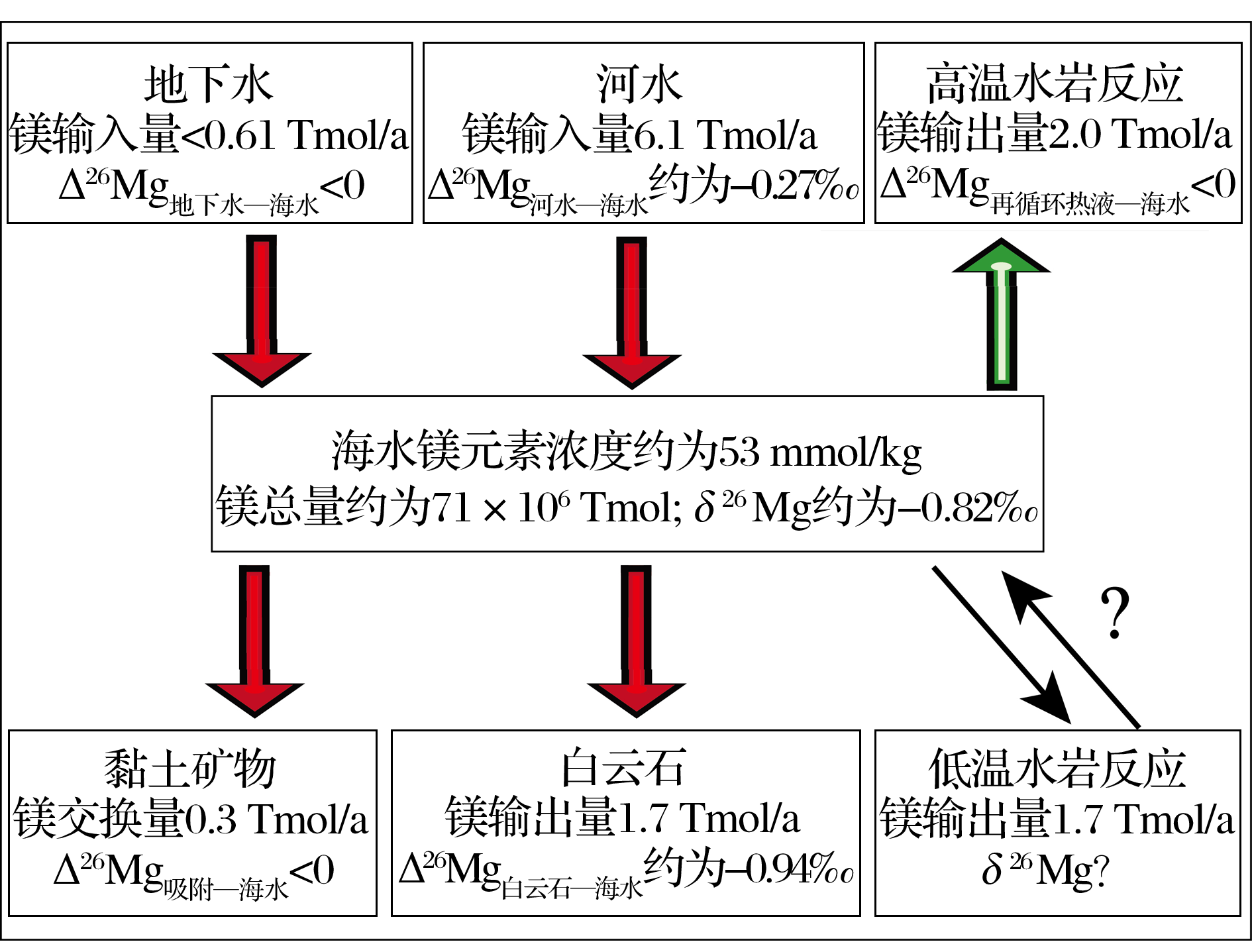 | 图5 海水镁同位素平衡示意图(镁的输送总量基于参考文献[100])Fig 5 A schematic Mg ocean isotope budget (Mg flux from reference[100]) |
由于河水的镁同位素组成比现代海水轻, 那么在目前较短的地质历史时期内T输入可能小于T输出.若基于Holland等[100]关于海洋镁通量的估算(J河流 = 6.1 × 1012 mol/a, J白云石 = 1.7 × 1012 mol/a), 那么T河流仍大于T白云石 (T河流 = (1.09-0.82)‰ × 6.1 × 1012 mol/a = 1.647 × 109 mol/a; T白云石 = (1.76-0.82)‰ × 1.7 × 1012 mol/a = 1.598 × 109 mol/a).假设洋中脊热液过程中不存在镁同位素的分馏 (T热液为0), 若T输入 ≤ T输出, 那么可能说明① 目前白云石的沉积总量被低估了; ② 仍存在其他过程导致轻的镁同位素从海水中移出(例如蒸发沉积过程中镁以水菱镁矿的形式沉淀)或温洋壳蚀变过程被低估等.若T输入 > T输出, 则可能说明海水的镁同位素组成逐渐变轻且向河水的方向演化.总之, 从目前的研究来看, 风化过程和碳酸盐沉淀过程仍是控制海水镁同位素组成的关键.水岩反应, 海水与黏土的离子交换, 近岸蒸发沉积, 生物作用等过程的镁同位素分馏及镁元素的迁移量需进一步的研究.
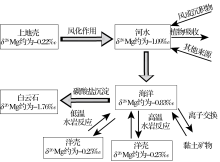
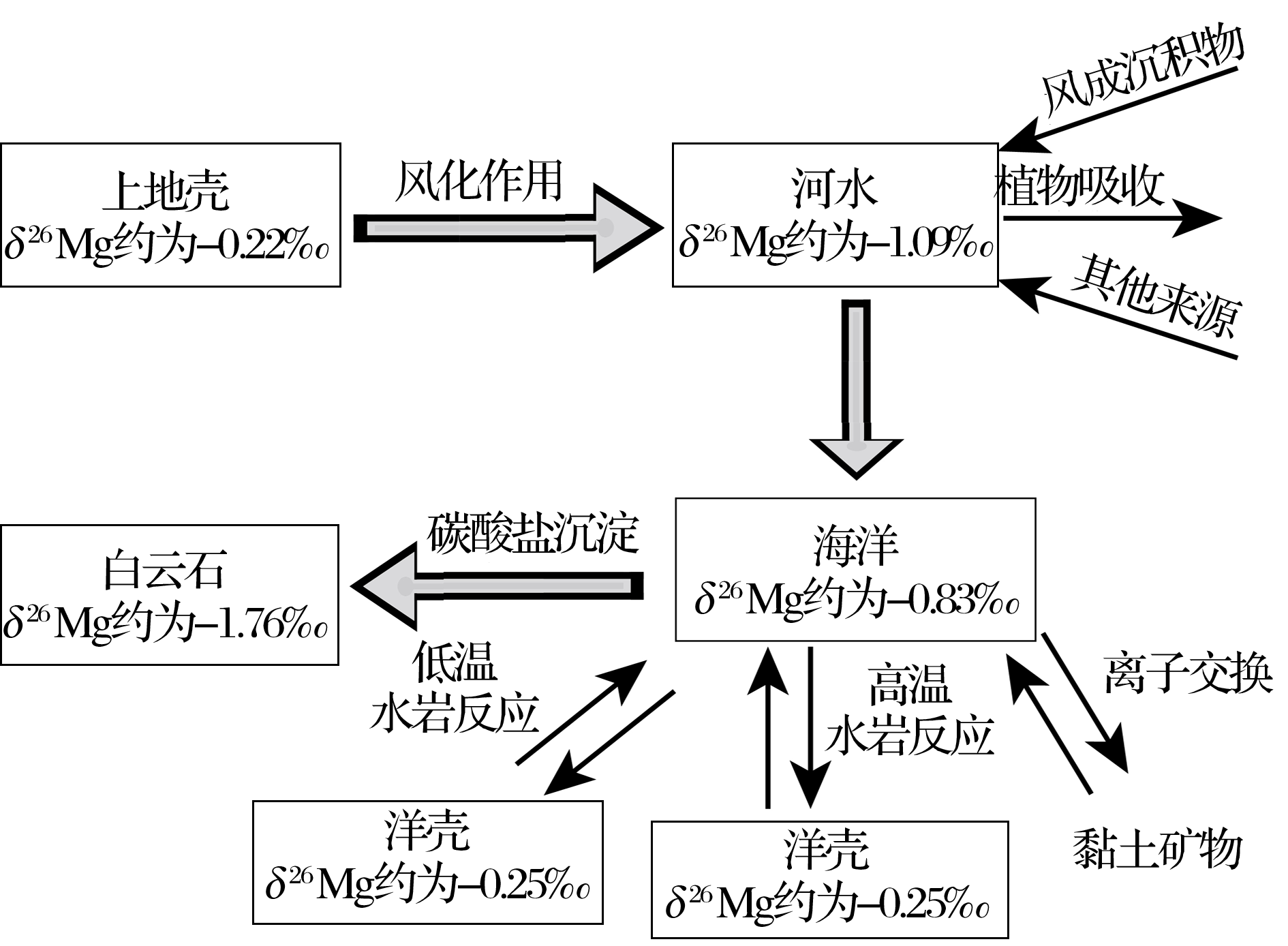 | 图6 表生环境中镁同位素地球化学循环示意图Fig 6 A schematic Mg isotope cycle in supergene environment |
表生环境中镁同位素地球化学循环概况见图6.上地壳岩石(δ 26Mg值平均约为-0.22‰ )经风化后, 镁以离子形式直接进入水体或以次生矿物的形式残留在原地.一般来说, 硅酸岩风化过程中镁同位素分馏主要受次生矿物影响.例如黏土矿物中富集重的镁同位素, 轻的镁同位素倾向于进入水体.进入水体的镁离子可经河流输送至海洋.在河流输送过程中, 镁同位素不发生分馏, 但河水的镁同位素组成易受外来物质输入(例如风成沉积等)等因素的影响.现代河水的镁同位素组成变化范围较大, 但总体上富集轻的镁同位素, 平均值约为-1.09‰ .现代海水的镁同位素组成均一(-0.82‰ ).海水中的镁离子以沉积碳酸盐岩的方式或在洋中脊发生水岩反应并被岩石吸收的形式发生迁移, 最终重新回到岩石中.碳酸盐岩沉积过程中镁同位素发生显著的分馏, 轻的镁同位素进入碳酸盐中.高温水岩反应也是镁离子被岩石吸收的主要过程, 此过程中可能发生镁同位素的分馏, 重的镁同位素可能倾向于进入岩石中.总之, 在表生环境中上地壳的镁(δ 26Mg值平均约为-0.22‰ )经历风化作用, 河流搬运, 海洋贮存, 最终以碳酸盐岩(镁同位素组成一般小于-1‰ )或与玄武岩发生反应的形式回到岩石圈.
火成岩具有相对均一的镁同位素组成(约为-0.25‰ )[38], 风化产物的镁同位素向变重的方向演化.相对于火成岩, 碳酸盐岩显著富集轻的镁同位素组成(一般来说小于-1‰ ).在岩浆过程, 变质脱水过程(绿片岩相, 角闪岩相, 麻粒岩相)镁同位素分馏非常有限[101, 102].因此, 镁同位素在岩石和矿床成因, 深部碳循环, 环境演化等方面具有一定的示踪潜力.限于篇幅, 这里仅对镁同位素示踪潜力作简要说明.
白云鄂博矿床的镁同位素研究为应用镁同位素示踪典型岩石和矿床成因提供了研究范例.白云鄂博矿床成因存在较大的争议, 争议的焦点之一是赋矿层白云质大理岩的成因, 观点主要集中在岩浆成因(火成碳酸岩)和沉积-交代成因, 还有学者认为是微晶丘.判断矿体镁质的来源是认识矿床成因的关键.最近的研究结果表明[26, 57]:火成碳酸岩墙样品的镁同位素组成主体与地幔橄榄岩接近; 赋矿白云岩的δ 26Mg变化范围为-1.13‰ ~ +0.30‰ , 整体变化范围较大, 主体与地幔橄榄岩接近; 而腮林忽洞微晶丘白云岩的镁同位素组成最轻, 约为-1.9‰ .从镁同位素角度来看, 矿体的镁不可能主要来自于沉积碳酸盐, 更倾向来源于岩浆且可能受到了上地壳物质和沉积碳酸盐的影响.因此, 整体上镁同位素结果支持火成碳酸岩成因模式.
对于深部碳循环而言, 镁同位素也具有一定的示踪潜力.在深部碳循环过程中, 火山作用释放的二氧化碳中无机碳约占95%, 可分为与俯冲相关的碳和原始幔源的碳, 且这些无机碳不能由传统碳同位素加以区分[103].地幔岩具有均一的镁同位素组成, 而大洋沉积碳酸盐岩的镁同位素组成显著轻于地幔岩.由于洋壳俯冲的脱水过程中全岩镁同位素不发生分馏[101], 那么以沉积碳酸盐岩形式进入地幔的镁就可以与原始幔源的镁加以区分.例如Yang等[19]发现华北克拉通中, 新生代玄武岩的镁同位素组成存在显著差异, 大于120 Ma的辽西义县组的玄武岩具有相对均一的镁同位素组成(-0.27‰ ± 0.05‰ ), 而小于110 Ma的阜新和太行山玄武岩的镁同位素组成具有较大的变化范围且整体偏轻(-0.46‰ ± 0.1‰ ).结合Sr, Nd, Pb同位素组成和元素地球化学特征, 发现小于110 Ma的阜新和太行山玄武岩具有亏损型地幔和类似地幔HIMU端元的特征.由于HIMU端元与俯冲洋壳再循环有关, 那么基于我国华北东部小于110 Ma的阜新和太行山玄武岩具有较轻镁同位素组成这一特征可推断其地幔岩浆源区混入了俯冲洋壳所携带的碳酸盐岩[19, 103].虽然镁同位素可以定性区分地幔来源的镁和洋壳俯冲所携带的沉积碳酸盐岩的镁, 但定量估算可能还需进一步的研究.首先地质历史时期沉积碳酸盐岩的镁同位素组成可能随着时间变化, 总体而言碳酸盐岩的镁同位素组成与海水的镁同位素组成密切相关.其次地质历史时期碳酸盐岩的主要类型也可能发生变化, 即以白云岩为主或灰岩为主等.因此, 应用镁同位素示踪深部碳循环方面还有很多问题需要研究.
目前关于应用镁同位素示踪古环境演化方面主要研究对象为黄土和盖帽碳酸盐岩.黄土是研究古环境演化的重要载体, 记录了新生代以来地球表生环境的演化历史.虽然全球黄土的镁同位素组成不均一, 但单一黄土剖面的镁同位素组成与风化程度密切相关.洛川黄土剖面的镁同位素组成与风化程度密切相关, 风化程度较强时镁同位素组成较重, 暗示了温暖湿润的气候; 风化程度较弱时镁同位素组成较轻, 暗示了干冷的气候[79].与黄土不同, 盖帽碳酸盐岩记录了地质历史时期海水的信息.Liu等[104]对澳大利亚和蒙古的盖帽碳酸盐岩进行了分步淋滤并测定了镁, 锶同位素组成, 发现基于镁, 锶同位素可将澳大利亚剖面的盖帽碳酸盐岩分为2类:一类具有较轻的镁同位素组成和较低的锶比值, 另一类具有较重的镁同位素组成和较高的锶比值且与蒙古剖面碳酸盐岩具有一致的镁, 锶同位素组成.第一类碳酸盐岩样品具有与沉淀碳酸盐岩相一致的锶同位素组成, 可能代表了其直接从海水中沉积.由于冰川融水(可能具有较重的镁同位素组成)导致海水分层, 提高了陆源输入的贡献, 进而导致沉积的白云岩也具有相对较重的锶, 镁同位素组成, 那么第二类样品可能表明了冰川融水对海水的显著影响.据此推测, 冰川融水与盖帽碳酸盐岩形成密切相关.
以上研究范例表明镁同位素能够对与碳酸(盐)岩有关的重要地质问题进行示踪.随着测试分析技术的发展以及镁同位素储库的完善, 镁同位素可望具有更广阔的示踪前景.
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
|
| [85] |
|
| [86] |
|
| [87] |
|
| [88] |
|
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
|
| [99] |
|
| [100] |
|
| [101] |
|
| [102] |
|
| [103] |
|
| [104] |
|