{kind=link}
{kind=link}
{kind=link}
Fe-Mo同位素与古海洋化学演化
引用本文
崔豪, 周炼, 李超, 彭兴芳, 金承胜, 石炜, 张子虎, 罗根明, 谢树成. Fe-Mo同位素与古海洋化学演化. 地球科学进展, 2013, 28(9): 1049-1056
Cui Hao, Zhou lian, Li Chao, Peng Xing fang, Jin Cheng sheng, Shi Wei, Zhang Zihu, Luo Gen ming, Xie Shu cheng. Iron - Molybdenum Isotopes and the Chemical Evolution of Ancient-Oceans. Advance in Earth Science, 2013, 28(9): 1049-1056
Permissions
Cui Hao, Zhou lian, Li Chao, Peng Xing fang, Jin Cheng sheng, Shi Wei, Zhang Zihu, Luo Gen ming, Xie Shu cheng. Iron - Molybdenum Isotopes and the Chemical Evolution of Ancient-Oceans. Advance in Earth Science, 2013, 28(9): 1049-1056
Fe-Mo同位素与古海洋化学演化
崔豪(1988-),男,湖北襄阳人,硕士研究生,主要从事古代与现代海洋生物地球化学研究. E-mail: ch_cuihao1@hotmail.com
摘要
Fe元素在自然界储量丰富, 而Mo元素则是海水中储量最丰富的过渡金属元素。由于Fe, Mo对其所在环境的氧化还原条件非常敏感, 近年来随着分析技术的进步, Fe与Mo同位素组成和变化被广泛用于鉴别古代海洋氧化还原状态及其演化。系统总结了古海洋化学研究中Fe与Mo同位素分馏机理和自然分布, 并对当前获得的FeMo同位素地史记录及其所指示的古海洋氧化还原状态进行了归纳与部分解释。造成Fe同位素分馏最明显的过程是氧化还原反应, 氧化态的Fe通常具有更重的Fe同位素组成;此外, 微生物作用及非生物作用下的黄铁矿生成过程也会产生明显的Fe同位素分馏。在海洋环境中Mo同位素的分馏主要与沉积物中铁锰(氢)氧化物吸附过程有关, 铁锰(氢)氧化物吸附Mo的过程中, 铁锰(氢)氧化物中倾向富集同位素较轻的Mo, 造成海水中Mo同位素偏重, 而硫化环境下的Mo沉积几乎不造成Mo同位素的分馏。Fe-Mo同位素的地史记录很好地说明了地质历史各时期海洋的氧化还原状态, 在2.3 Ga以前海洋主要为铁化的状态, 其中在2.6~2.5 Ga时氧含量略有增加;2.3~1.8 Ga之间地球表面初步氧化, 硫化物沉积增加;1.8~0.8 Ga时海洋中的硫化环境得到了扩张;0.8 Ga以后地球表层逐步氧化, 硫化水体消退。最后, 对古海洋化学研究中FeMo同位素研究未来的工作重点给予了展望。
关键词:
Fe同位素; Mo同位素; 氧化还原状态; 古海洋化学演化
中图分类号:P736.4
文献标志码:A
文章编号:1001-8166(2013)09-1049-08
Iron - Molybdenum Isotopes and the Chemical Evolution of Ancient-Oceans
Abstract
Iron(Fe) is abundant in nature while molybdenum(Mo) is the most abundant transition metal in seawater. Due to their high sensitivity to the redox state of the environment, the isotopic compositions of Fe and Mo as well as variations have been widely used to probe the redox conditions and the evolution of ancient ocean chemistry in favor of improved analytical techniques. Here, we summarized isotopic fractionation mechanisms and natural distribution of both iron and molybdenum isotopes, and further we summarized and partially reinterpreted the redox evolution of ancient oceans through time based on available FeMo data compiled in this study. The process that causes the largest iron isotope fractionation is redox reaction and the iron in oxidation state is generally enriched in56Fe. Biotic and abiotic pyrite formations also produce a large Fe isotope fractionations. Isotopic fractionation of molybdenum in seawater is mainly caused by the adsorption process of dissolved Mo onto ferromanganese oxides or hydroxides in sediments. FeMn (hydro)oxides tend to adsorb isotopically light molybdenum resulting in the isotopic composition of Mo in seawater heavier. However, the Mo sinks in euxinic settings cause almost no molybdenum isotope fractionation. The Fe-Mo isotope isotopic records through geological timegenerally suggest similar ocean redox evolution: Oceans older than 2.3 Ga was mainly dominated by ferruginous condition, and there was a slight increase in oxygen content between 2.6 and 2.5 Ga. Earth’s surface was initially oxidized during 2.3 to 1.8 Ga, during which euxinic deposition of sulfide was elevated. Euxinic waters may have expanded greatly between 1.8 and 0.8 Ga, and after that, Earth’s surface had being gradually oxidized and the euxinic waters shrank substantially.Finally, suggestions are proposed for further work on the FeMo isotope research in the context of ancient ocean chemistry.
Keyword:
Iron isotope; Molybdenum isotope; Redox state; Ancient ocean chemistry.
1 引言
越来越多的研究表明地球生命的起源和演化与地球表层环境, 特别是古代海洋的氧化还原状态密切相关[ 1, 2, 3]。在过去的几十年中, 古海洋氧化还原化学的研究手段日渐丰富, 主要包括Fe-S-C化学、微量元素化学、铁钼同位素等手段[ 4]。其中, 铁与钼同位素组成(分别为δ56Fe和 δ98/95Mo或δ97/95Mo)作为近年来新兴的研究手段, 在古海洋氧化还原化学的研究中逐渐显示了其独特的应用潜力。
铁元素在地壳中含量丰富, 由于其过渡元素的特征使得铁元素成为地球上参与氧化还原化学过程最广泛的元素之一[ 5]。而由于钼酸盐极好的稳定性和可溶性, 钼元素则是今天海水中含量最高的过渡金属元素[ 4]。铁钼元素的共同特点是:二者都对不同氧化还原状态反应敏感, 且在不同的氧化还原状态下, 以不同的价态及化合物的形式存在并具有不同的同位素组成。这一特点使二者的同位素组成成为沉积水体的氧化还原状态的潜在指示剂[ 4]。然而, 由于铁、钼元素质量数较大, 相比其他轻同位素而言, 其同位素变化不明显, 铁钼同位素测定精度成为这一手段能够被广泛应用的关键。近年来, 多接收杯电感耦合等离子体质谱仪(MC-ICP-MS)分析方法的改进及测试精度的提高为铁钼同位素组成的精确测定提供了可能, 也为铁钼同位素方法在古海洋化学研究中的广泛应用奠定了基础[ 6, 7, 8]。
2 计量方法
常用δ56Fe = { [ (56Fe/54FeSample) / (56Fe/54FeStandard)-1 ] ×103 }来表示铁同位素组成[ 9]。火成岩的铁同位素组成(56Fe/54Fe≈0‰)基本不变, 通常被当作计算δ56Fe的标准[ 9, 10], 也有许多学者用IRMM-014(一种国际同位素标样) 作为标准。
常用δ98/95Mo = { [ (98Mo/95MoSample) / (98Mo/95MoStandard)-1 ] ×103 }或者δ97/95Mo={ [ (97Mo/95MoSample) / (97Mo/95MoStandard)-1 ] ×103}来表示钼同位素组成, 其中δ97/95Mo≈2/3·δ98/95Mo[ 11]。标准通常使用JMC(Johnson Mattey公司制作的标准溶液)或者平均大洋水(δ98Mo/95Mo≈2.3‰)。
3 分馏机理与自然分布
3.1 Fe同位素
在低温系统中, 铁同位素组成最重要的控制因素是氧化还原反应—氧化态通常比还原态或中间态更富集重同位素, 因此, 通常Fe3+有着较高的δ56Fe值而Fe2+有着较低的δ56Fe值[ 12]。在现代富氧大气条件下, 虽然陆地风化产物中Fe3+/Fe2+的比例很高, 但是由于Fe3+极低的溶解度, 通过液相流失的Fe3+微不足道[ 13]。然而, 由于现代海洋相对于古代海洋来说总体贫Fe2+, 所以通过地表径流进入海洋的少量同位素较轻的溶解铁仍可以对海洋溶解铁同位素组成产生重大影响[ 14]。在浅水区域, Fe2+会被迅速氧化(通过有氧或者无氧氧化)而产生同位素较重的Fe3+沉淀和同位素相对较轻的溶解Fe2+[ 15, 16]。此外, 铁(氢)氧化物对溶液中溶解铁的吸附作用会优先吸附同位素较重的铁[ 17], 导致液相Fe2+有变轻的趋势。
大陆架区域有着丰富的微生物作用, 其可以产生明显的铁同位素分馏。异化铁还原过程(Dissimilatory Iron Reduction, DIR)将优先还原铁氧化物或氢氧化物中同位素较轻的成分, 可产生比还原前铁(氢)氧化物δ56Fe值低1‰~ 2‰的Fe2+aq[ 13, 17~ 20]。由于细菌硫酸盐还原过程(BSR:bacterial sulfate reduction)所形成的H2S将优先与同位素较轻的铁反应形成黄铁矿, 这一过程使剩余的Fe2+aq略微变重而形成的黄铁矿同位素较轻[ 21]。然而, 最近的一项研究表明:在非生物作用下, Fe2+形成黄铁矿的过程也可以产生大范围的铁同位素分馏(+1.7‰ ~ +3.0‰)[ 22]。
一些储库及过程中铁同位素组成范围见表1。
| 表1 一些过程及储库中铁同位素组成范围 Table1 Isotopic composition of Fe in some processes and reserviors |
3.2 Mo同位素
现代海洋中, Mo主要以可溶的钼酸盐(MoO42-)形式存在, 其同位素组成十分均一。在海洋环境中Mo同位素的分馏主要与沉积物中铁锰(氢)氧化物吸附过程有关。铁锰(氢)氧化物吸附Mo的过程中, 铁锰(氢)氧化物中倾向富集同位素较轻的Mo, 造成海水中Mo同位素偏重, 以致海水和铁锰(氢)氧化物之间的分馏可达2‰。基于相同的原理, 现代风化过程中, 轻的同位素优先吸附于土壤中, 尤其是铁锰(氢)氧化物表面, 致使河流输入海洋的Mo同位素偏重, 平均0.7‰[ 27]。在硫化环境([H2S]>11μM)下, 海水中的Mo几乎全部以颗粒硫化物(MoS42-)的形式迅速沉积下来, 所产生的Mo分馏极小, 导致沉积物中Mo同位素值与海水中Mo同位素值大致相当[ 4]。因此, 硫化环境下沉积物中的Mo同位素组成可以用来追踪沉积时海水的Mo同位素组成。而当[H2S]介于0.01~11μM时, Mo的形态分别为MoO42-、MoO4-xSx2-和MoS42-, 上述不同形态Mo进入沉积物中的比例使得Mo同位素分馏存在较大的差异, 目前, 此环境下Mo同位素的分馏特征还不是十分清楚。最近, Scott等人[ 28]对非硫化环境沉积物中Mo循环及其同位素分馏特征进行了总结, 认为该沉积环境下Mo同位素组成也能够较好的示踪古氧化还原状态, 主要存在两种情况:①不存在Mo氧化循环并且海水中的H2S能够使得MoO42-定量的转变为MoS42-, 此时沉积物Mo同位素值能够代表海水中Mo同位素组成;②非硫化沉积物孔隙水中, 由于锰氧化物优先吸附轻同位素馏分的MoO42-, 导致孔隙水中Mo同位素组成越来越轻, 并能够保存在深部硫化带中。综上所述, 海水中Mo同位素组成能够较好地指示水体沉积时的氧化还原条件。一些储库中Mo同位素组成范围见表2。
| 表2 不同储库里Mo同位素组成范围 Table2 Isotopic composition of Mo in some reserviors |
4 地质记录与古海洋化学演化
4.1 Fe同位素的地史记录
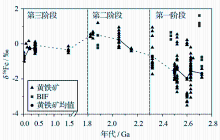
图1为地质历史时期沉积黄铁矿与条带含铁建造(Banded Iron Formation , BIF)沉积中Fe同位素的地史记录。依据Fe同位素组成特征, 我们将将古海洋氧化还原化学演化分为3个阶段。
第一阶段为2.3Ga以前, 这个阶段黑色页岩中黄铁矿的Fe同位素组成有着较大的变化范围(+0.5‰ ~ -3.5‰), 并且最低(平均为-1.49‰)。国内外学者对这一现象有不同的看法。Icopini等[ 17]提出, 铁氧化物微粒可能优先吸附溶解铁中同位素较重的铁, 因此可以导致溶液中残余铁同位素较负并被记录在沉积黄铁矿中。Rouxel等[ 23]据此进一步认为此阶段黄铁矿较轻的铁同位素组成可能反映了黄铁矿中Fe2+的热液来源和铁氧化物沉淀对同位素组成较重铁吸附的共同作用。此阶段条带含铁建造中较重的铁同位素组成支持了此观点(见图-1)。Severmann等[ 33]对氧化-次氧化大陆架沉积物孔隙水中的铁同位素组成进行了研究, 结果表明:含有铁(氢)氧化物的孔隙水可以通过DIR过程产生同位素较轻的Fe2+aq, 由此, 经历了强烈次氧化铁循环的大陆边缘沉积物可能存在着一个低δ56Fe值的海底Fe2+aq通量。Severmann等[ 35]对黑海沉积物中铁同位素组成做了进一步研究, 表明从大陆架到深部盆地的底层铁传输, Fe2+经历了复杂的氧化还原过程后可使深部海水的δ56Fe值降低到-1‰ ~ -2‰;此外, 硫化物限制的条件下形成的黄铁矿与海水中的Fe2+aq间也可产生约-1.5‰的分馏[ 23, 36], 这样就可以形成δ56 Fe范围为-2.5‰ ~ -3.5‰的黄铁矿, 从而解释了太古代具有极低δ56Fe值黄铁矿的成因。然而, 需要指出的是在微生物活动很频繁的现代缺氧沉积物中却没有观察到如此大的铁同位素分馏[ 33, 37]。Guilbaud等[ 22]近来对此又提出了新的看法:在非生物作用下, Fe2+形成黄铁矿的过程也可以产生极大的铁同位素分馏(+1.7‰ ~ +3.0‰)。由于自然界黄铁矿的同位素组成都落在非生物成因的黄铁矿形成过程所产生的分馏范围, 由此, 太古代铁同位素的极端负值并不一定由氧化还原和DIR过程形成。这一研究将铁同位素在追踪古海洋氧化还原状态的应用复杂化。Anbar等[ 32]指出当高浓度溶解铁在缺氧低硫化条件下由于部分铁氧化与部分铁还原可以产生很大的δ56Fe变化, 可以较好解释此阶段铁同位素组成的巨大波动。
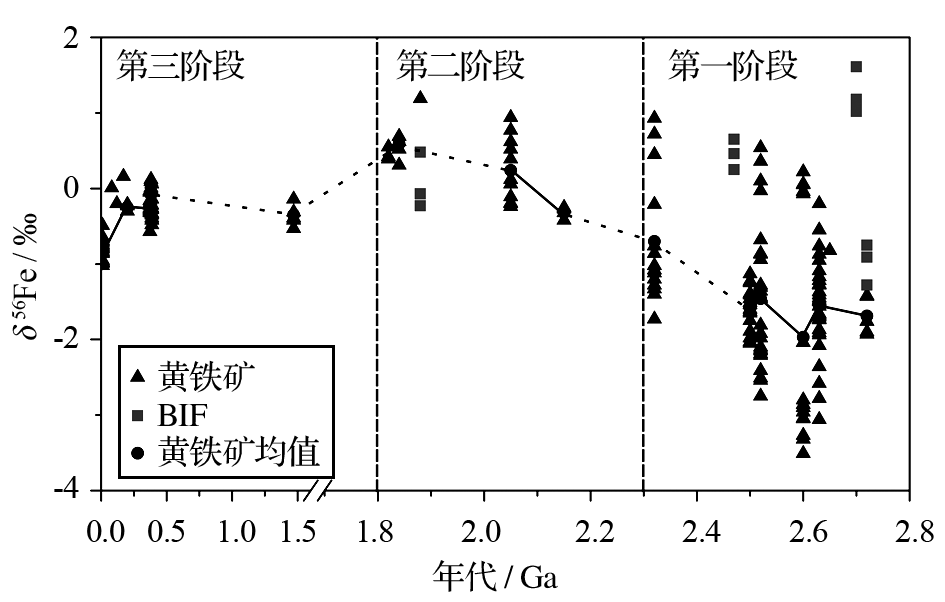 | 图1 沉积黄铁矿与条带铁矿建造(BIF)中所记录的Fe同位素组成的地史记录数据来源:近代来自文献[33];中晚泥盆来自文献[34];其余数据汇编来自文献[23];均值计算依据黄铁矿中铁同位素组成。Fig. 1 Historical fluctuation of Fe isotopes recorded in sedimentary pyrites and BIFsData sources: Modern from reference[33]; Mid-late Devonian from reference[34]; Others from reference[23]; The mean is the average of the iron isotopic compositions in pyrite |
综上所述, 尽管数据的解释上仍存在着很大的不确定性, 太古代沉积黄铁矿铁同位素组成基本确定了当时的沉积环境为缺氧铁化(含游离的Fe2+)。这与此时极低的大气氧含量和广泛存在的BIF沉积等背景[ 38]相符。
第二阶段为2.3Ga到1.8Ga, 此阶段黑色页岩中黄铁矿的δ56Fe负值逐渐消失并出现高达+1.2‰的正值。有大量证据表明2.3Ga时大气氧含量已明显增加(即古元古代大氧化事件[ 39])。一般认为:古元古代随着大气氧含量的增加, 地表环境逐步氧化, 大陆风化作用的增强使河流输入海洋的硫酸盐增加, 进一步导致海洋硫化水体和黄铁矿沉积的增加。增加的黄铁矿沉积可能逐步耗尽了海洋中同位素较轻的铁而使海洋溶解铁库同位素组成逐步变重[ 32, 40]。我们认为还有一种可能:随着氧化风化导致陆地(氢)氧化铁的增加, 通过大气沉降、河流与冰川搬运的同位素较重的颗粒氧化铁也极大增加, 这样, 这些同位素较重的颗粒氧化铁在缺氧还原海洋, 特别是化学跃变层中被还原为Fe2+, 为近岸硫化水体[ 38, 41]中黄铁矿的形成提供了重要的铁源, 从而导致地质记录中黄铁矿的铁同位素组成逐步增加。
第三阶段1.8Ga至显生宙, 此阶段数据较少, 但可得的黄铁矿δ56Fe变化不大, 范围为0‰到-1‰。在Fe2+aq含量高而缺氧低硫化物的环境下, 由于部分铁被氧化与部分铁被还原, 将产生大的铁同位素范围, Rouxel等[ 23]据此提出假设:沉积黄铁矿中的铁同位素变化范围对海水中溶解铁含量很敏感, 溶解铁越多则δ56Fe变化越大。由此说明第一阶段到第三阶段逐渐缩小的铁同位素数值的变化范围可能反映了海洋中溶解铁储量的减少[ 32]。相对其他两个阶段, 第三阶段海洋广泛的缺铁与BIF沉积在1.8Ga的基本消失(除晚新元古代冰期时期BIF短暂出现)相符合。
4.2 铁同位素的空间变化
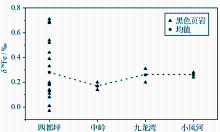
目前关于地史时期铁同位素空间差异性的研究甚少, 对这一主题研究的一个重要实例来自研究程度较高的新元古代埃迪卡拉纪陡山沱组[ 42]。样品采自湖北宜昌小风河(浅水台地相)、九龙湾(内陆架)、湖南石门中岭(陆地边缘)和张家界四都坪(斜坡相)4个剖面陡山沱组底部盖帽白云岩之上的二段底部黑色页岩。古地理研究表明陡山沱组二段底部黑色页岩沉积于开阔海洋状态[ 43]。图2为4个不同剖面的陡山沱组二段黑色页岩全岩的δ56Fe数据。
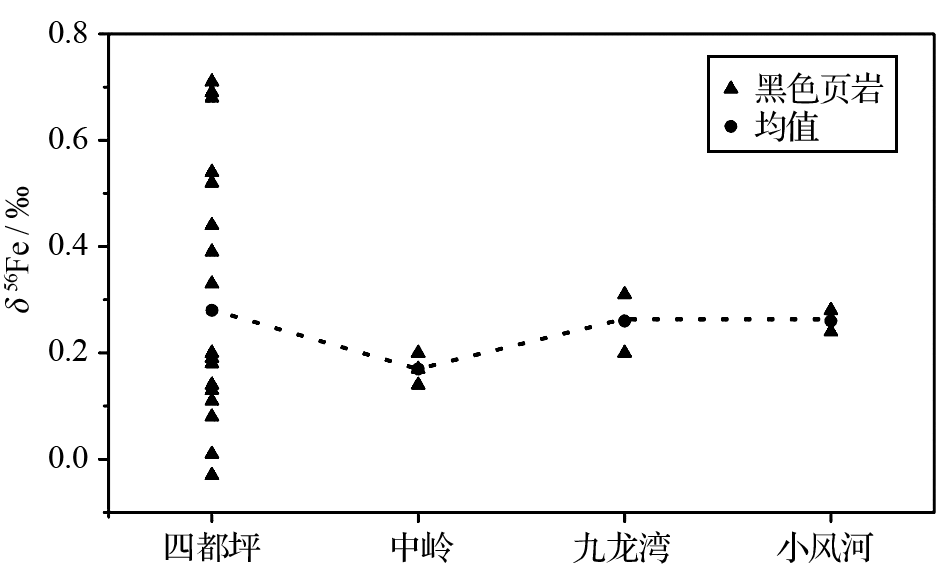 | 图2 新元古代埃迪卡拉纪陡山沱组二段黑色页岩全岩的δ56Fe数据的空间分布。沉积相:宜昌小风河为浅水台地相;九龙湾为内陆架;湖南石门中岭为陆地边缘;张家界四都坪为斜坡相Fig. 2 The spatial distribution of whole rock δ56Fe data from the black shales of MemberⅡof the Ediacaran Doushantuo FormationSedimental faces: Xiaofenghe is platform; Jiulongwan is Inner shelf; Zhongling is Shelf margin; Siduping is Slope |
从图2可以看出, 四都坪剖面的δ56Fe值变化较大。根据新元古代新近提出的“三明治”海洋结构模型[ 3], 此时大陆架斜坡处硫酸盐含量很低, 在缺氧低硫的环境下, 由于部分铁氧化与部分铁还原, 可以形成较大范围的铁同位素分馏[ 32]。中岭地区此阶段铁组分化学与硫同位素数据均表明其处于近岸硫化水体中[ 3], 细菌硫酸盐还原(BSR)过程强烈, 岩石中黄铁矿含量高, 而BSR形成的H2S将优先反应同位素轻的Fe2+来形成黄铁矿, 由此可以解释中岭地区为什么δ56Fe值相对其他地区较低。我们不能对目前有限的数据做过分的解读, 然而, 当前的有限的数据的确能表明古海洋的氧化还原状态, 至少在新元古代陡山沱时期存在着重要的空间差异性。
4.3 Mo同位素的地史记录
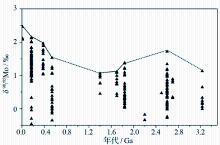
由于氧化水体与硫化水体面积的相对多少决定了海水中Mo同位素组成, 测定地质历史时期硫化页岩中的Mo同位素组成便可以有效重建地球海洋化学演化过程中硫化水体面积的相对变化[ 28, 52]。由于在高度硫化的水体中([H2Saq]>11μM)海水中的钼酸盐几乎能够完全转换成活性的MoS42-并与有机质反应而沉积, 此过程几乎不发生Mo的同位素分馏;而黑色页岩多沉积于缺氧和硫化的状态下, 所以其全岩的Mo同位素组成的最大值最有可能代表了沉积时期海水Mo同位素组成。图-3概括了目前可得的黑色页岩全岩的Mo同位素地史数据, 其显示了多阶段的演化特征, 现分述如下:①早期地球(>2.8Ga)海洋Mo同位素数据较少, 目前可得的只有来自32亿年南非Fig Tree Group的研究[ 43]。虽然我们不能排除此阶段的数据有可能反映当时海水的Mo同位素组成, 但由于大气和海洋均处于还原状态, 海洋中以氧化状态的MoO42-含量极低[ 53], 我们认为海洋沉积物中的Mo几乎全部来自陆源碎屑而非硫化海水自生沉积, 因此, 此阶段全岩中的Mo同位素组成应该反映的是陆源碎屑Mo同位素组成。目前已知最早的海水硫化是来自西澳大利亚晚太古代Jerrinah组(2.66Ga)[ 53], 早于此的海水硫化尚未见报道。②晚太古代(2.8-2.5Ga), 大气氧气含量已出现小幅增加, 陆地Mo已能够被氧化成MoO42-[ 31, 54], 在海洋中已局部存在硫化环境[ 53, 54], 因此, 黑色页岩能够记录当时海洋Mo同位素组成。在2.6-2.5Ga之间, 海洋Mo同位素组成曾经高达1.86‰[ 55], 这可能由于铁-锰氧化物吸附了同位素轻的Mo而使海水的Mo同位素组成变重, 最终在缺氧硫化环境下沉积的黑色页岩中得以记录。因此, 这有可能反映了在大氧化事件之前就出现了大气氧含量的增加[ 44, 55], 使得Mo同位素组成(1.86‰)接近于现今海洋[ 55]。③古元古代早-中期(2.5-1.8Ga)。自古元古代早期, 地球经历了大氧化事件(GOE, 约2.4Ga)和Lomagundi事件(约2.1Ga), 生物圈的氧化程度明显得到增强, 大量的陆源风化Mo输入海洋, 海洋中Mo库迅速增加。然而, 目前该时期的Mo同位素组成数据很少, 我们无法据此对当时的海洋化学状态做过多的探讨。④古元古代晚期-新元古代早期(1.8-0.8Ga), 数据也相对有限, 只有18.4亿年的Rove组[ 51]、17亿年的Wollogorang组和14亿年的Velkerri组[ 45]。有限的数据显示此阶段海水中Mo同位素组成较今天的海洋(+2.4‰)明显要低。古元古代晚期至新元古代早期大气氧含量虽然低于现今, 但已得到明显增加。陆源风化来源的硫酸盐对海洋的输入因此得到提高并导致硫化水体的广泛扩张[ 56], 由此促使海水中的Mo主要以MoS42-的形式在硫化环境中沉积, 而氧化海水中铁锰氧化物对海水Mo吸附比例下降, 从而导致该时期海水Mo同位素组成明显低于现今海洋[ 11, 51]。⑤新元古代中晚期-现代(<0.8Ga), 海洋Mo同位素组成显示了持续增加并到达现今海洋Mo同位素组成。这一特征与页岩中Mo的高度富集反映了地球表层自晚新元古代以来的极大氧化所导致的海洋缺氧水体的迅速消退和Mo库的迅速增加[ 57, 58]。海洋的极大氧化使得铁锰氧化物大量沉积, 由于铁锰氧化物易于富集同位素较轻的Mo, 促使同位素较轻的Mo优先沉淀出来, 从而导致海洋中Mo同位素组成变重。
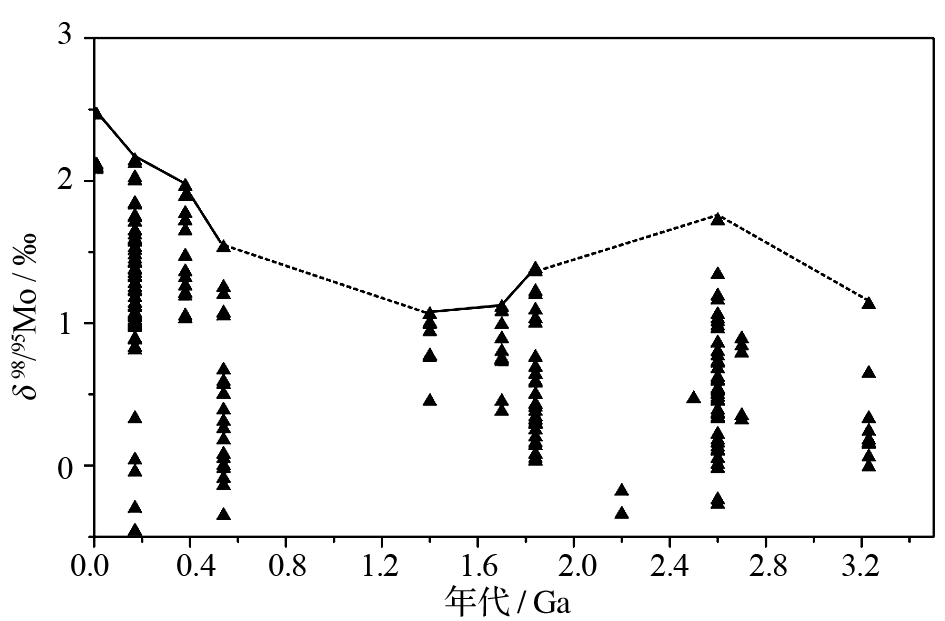 | 图3 黑色页岩全岩所记录的Mo同位素组成的地史波动(数据来源:文献[11, 44~51])Fig 3 Historical fluctuations of bulk Mo isotopes recorded in black shales(Data sources: references [11, 44~51]) |
5 结论与展望
控制铁同位素组成的最重要的因素是氧化还原反应, 氧化态的铁通常富集同位素较重的组分, 而还原态的铁则富集同位素较轻的组分。目前已知的可以引起铁同位素分馏的过程有:氧化还原变化、异化铁还原(DIR)、细菌硫酸盐还原(BSR, 虽然这个过程是对硫酸根的还原, 但是生成的硫化氢将优先与同位素轻的溶解铁反应形成黄铁矿)和铁(氢)氧化物的吸附作用。钼同位素的分馏则主要发生在铁-锰氧化物对同位素较轻Mo组分的偏好吸附, 而在较强硫化沉积过程中钼同位素几乎不发生分馏。
Fe同位素的地史波动将海洋化学演化分为3个阶段。第一阶段为2.3Ga以前, 沉积黄铁矿的δ56Fe值较负并且伴随着大的波动, 反映了缺氧低硫的铁化环境;第二阶段为2.3~1.8Ga, δ56Fe的强烈的负值消失, 出现正值并且变化范围缩小, 反映了地球表面环境的初步氧化和硫化物沉积的增加;第三阶段为1.8Ga以后, δ56Fe值的变化范围被进一步缩小, 反映了大气及海洋氧含量的增加, 海洋中溶解铁含量的减少。
Mo同位素的地史记录所反映的古海洋化学演化与Fe同位素大致相当, 但更为详细。2.8Ga以前大气和海洋都处于还原状态, 海洋沉积物中的Mo同位素组成应该反映陆源碎屑Mo的同位素特征;在大氧化事件之前(2.6~2.5Ga之间)大气氧气含量就有增加;中元古代时期(1.8~0.8Ga), Mo同位素组成明显低于现今海洋, 表明了硫化环境的扩张;晚新元古代-现代, 海洋Mo同位素组成的持续增加并逐步达到今天海洋状况反映了大气—海洋逐步氧化和海洋硫化水体的消退。
总的来说, 钼同位素的地史记录与铁同位素的地史记录相结合, 共同反映了地球表层逐步氧化的过程, 但显然目前对钼同位素形成机制的认识和数据记录的解释更为成熟, 而铁同位素手段在数据解释上还存在较大争议。对于铁同位素, 还有许多地方需要进一步研究, 比如不同矿物相与流体相及矿物之间的铁同位素分馏系数、各种反应及过程(如异化铁还原、微生物硫酸盐还原及非生物黄铁矿形成)的铁同位素分馏机制等。
此外, 前人对于铁—钼同位素的研究多涉及时间记录的波动, 而对其其空间的波动则很少关注。目前的古海洋化学结构模型显示古海洋化学存在着广泛的空间差异性[ 3, 37]。因此, 测量及研究海洋不同深度沉积物的铁—钼同位素数据对于发展和完善古海洋化学中空间结构演化具有重要意义。最后, 如果把铁-钼同位素手段与其他古海洋化学研究手段如硫、碳同位素、铁化学、微量元素等结合, 相互补充, 将能更加精确地说明整个地球海洋的化学演化历史。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|